
Lignée granuleuse
Ce chapitre sur la ligné granuleuse est divisé en 6 parties :
(le chapitre 4 - Leucémies aiguës myéloïdes a été fractionné en deux pour optimiser le temps de chargement des photos)
5 - Myélodysplasies
Les myélodysplasies forment un ensemble complexe de maladies dont l'unicité est dans l'atteinte des 3 lignées médullaires responsable d'une pancytopénie à moelle riche : cytopénie périphérique avec anomalies morphologiques des éléments sanguins et moelle riche, plus ou moins dystrophique (insuffisance médullaire qualitative). Il existe quelques rares myélodysplasies congénitales mais la plupart des myélodysplasies sont acquises, primitives ou secondaires.
1 – Diagnostic cytologique des myélodysplasies
Quelqu'en soit la cause les myélodysplasies comportent plus ou moins les mêmes anomalies sanguines et médullaires. Toutes les anomalies ne coexistent pas en même temps, elles apparaissent le plus souvent progressivement et se complètent. N'importe quelle anomalie peut inaugurer la maladie, mais la plus fréquente porte sur les globules rouges avec une anémie. Au total le diagnostic de myélodysplasie, suggéré par les chiffres de la NFS, se précise par l'examen attentif de la lame de sang à la recherche de la moindre anomalie morphologique évocatrice, il y a une « atmosphère » myélodysplasique. Dans nombre de cas il faudra compléter la démarche diagnostique par une ponction de moelle et les dystrophies constatées des différentes lignées viendront fortement étayer le diagnostic.
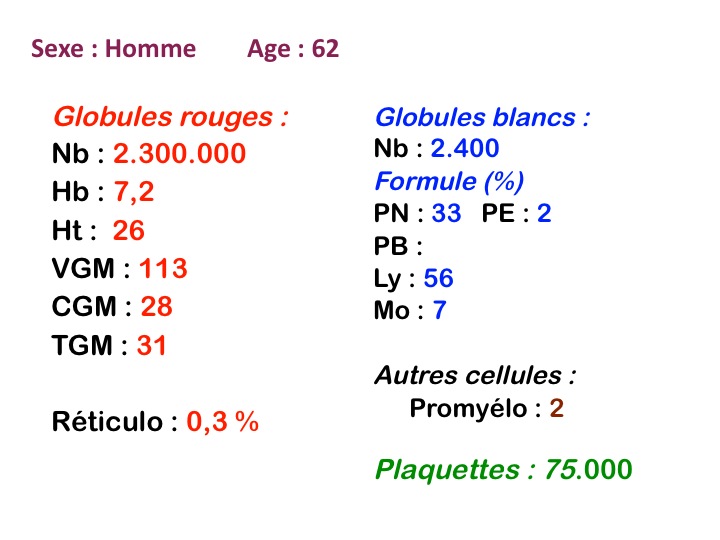
Les manifestations sanguines des myélodysplasies portent sur l'ensemble des éléments sanguins d'origine médullaire, mais le tableau hématologique se complète progressivement.
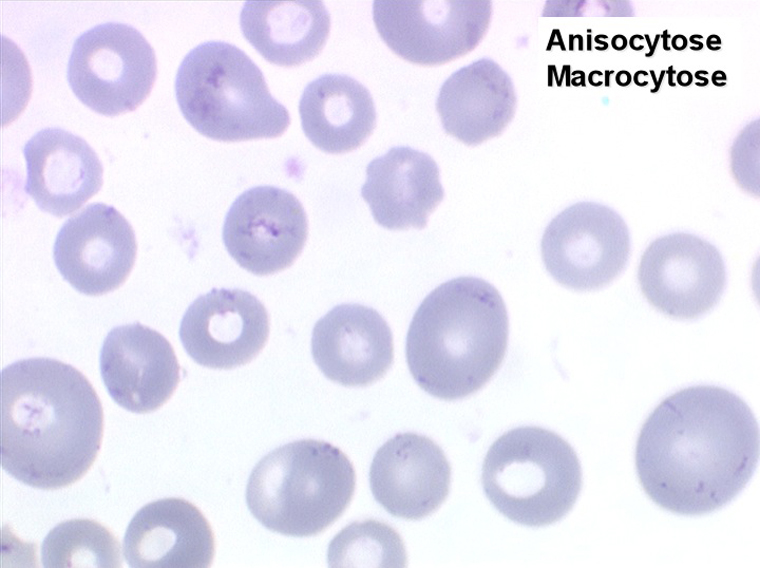
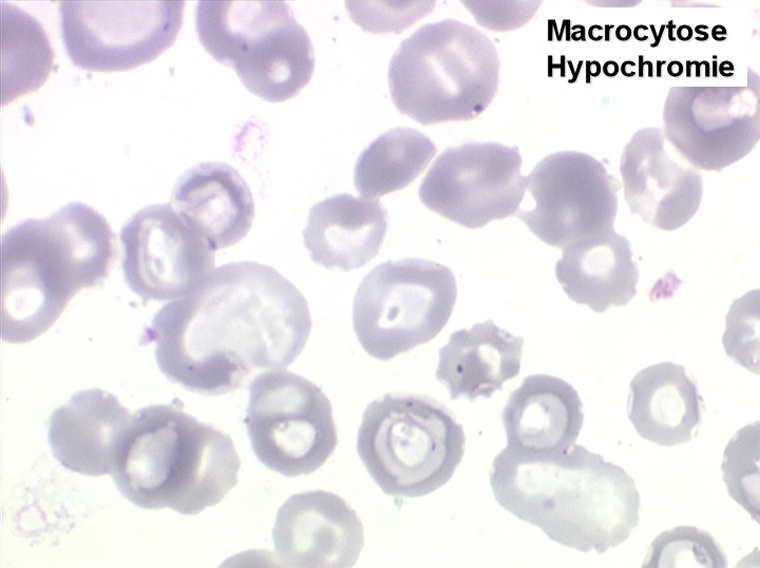
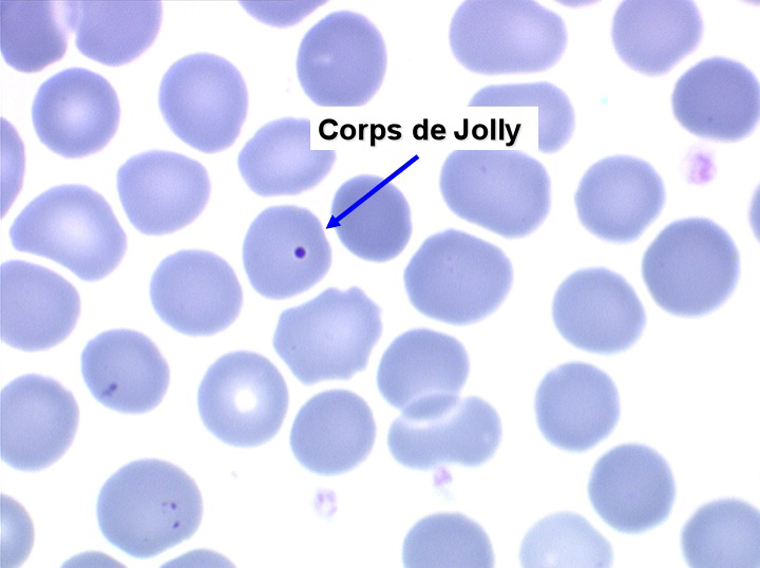
1. L'anémie est le signe le plus fréquent et le plus précoce, elle est non ou peu régénérative, habituellement macrocytaire. L'association d'une macrocytose et d'une hypochromie est particulièrement évocatrice de myélodysplasie. Les déformations des hématies sont variées : anisocytose, poïkylocytose, schizocytose, dacryocytose, ovalocytose. Il est non moins fréquent d'observer dans les hématies des inclusions : corps de Jolly, anneaux de Cabot, ponctuations basophiles.
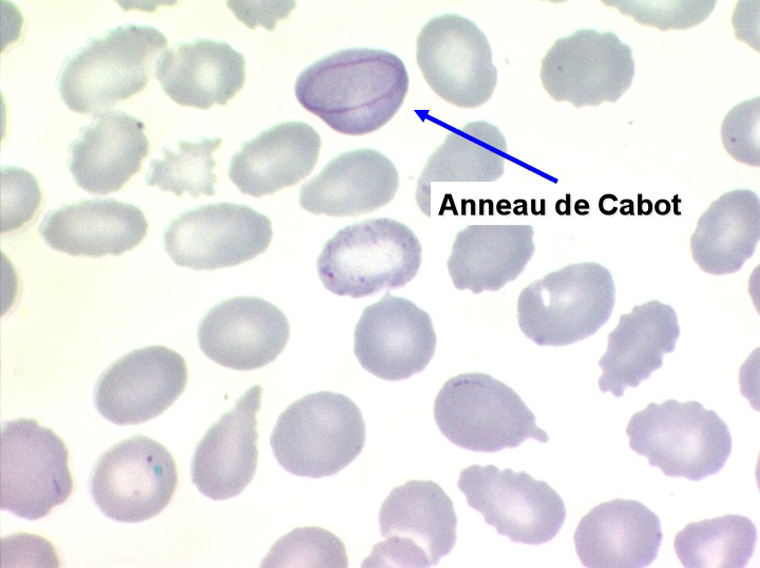
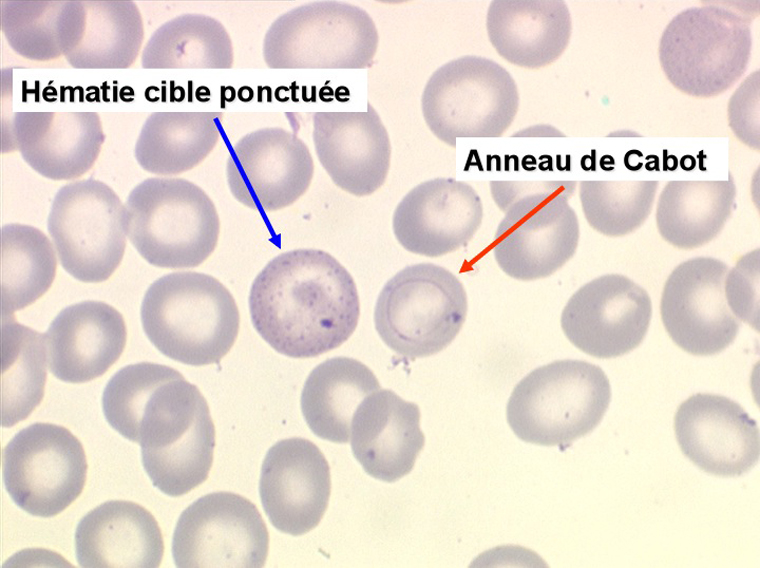
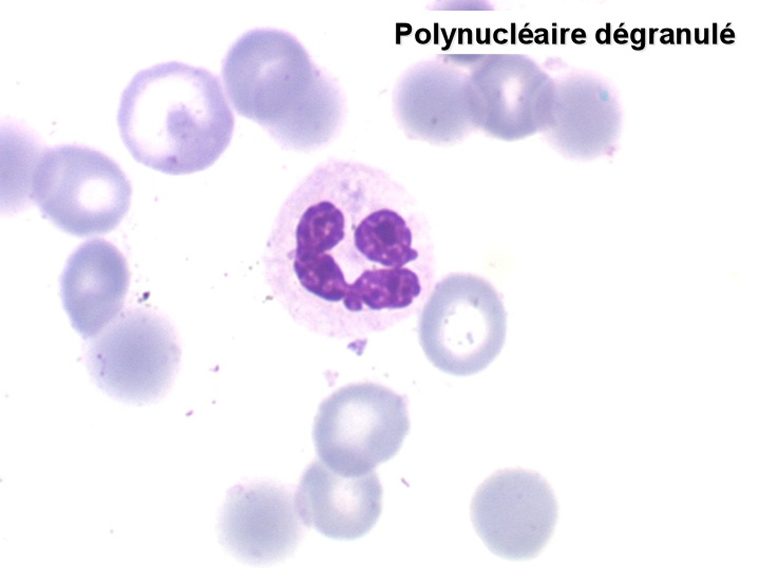
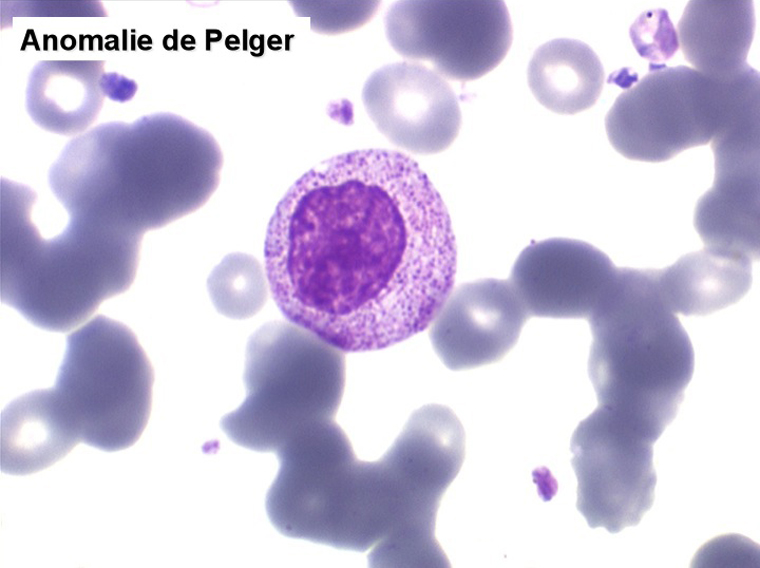
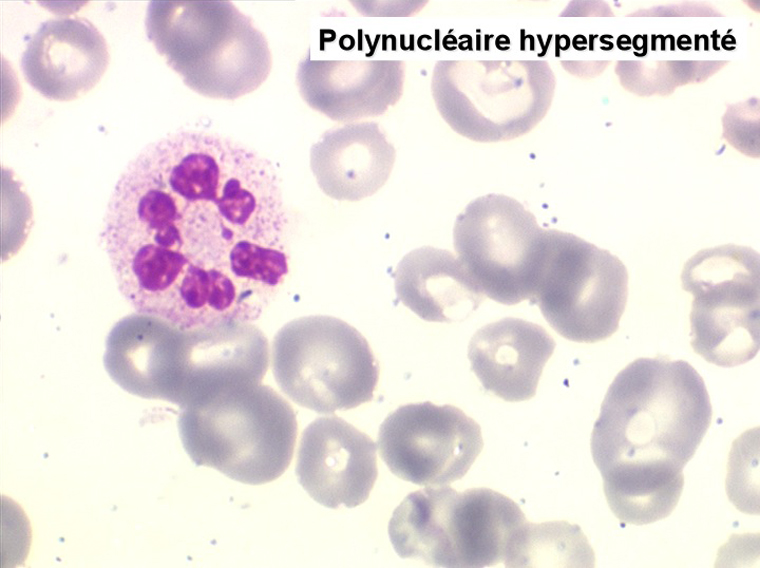
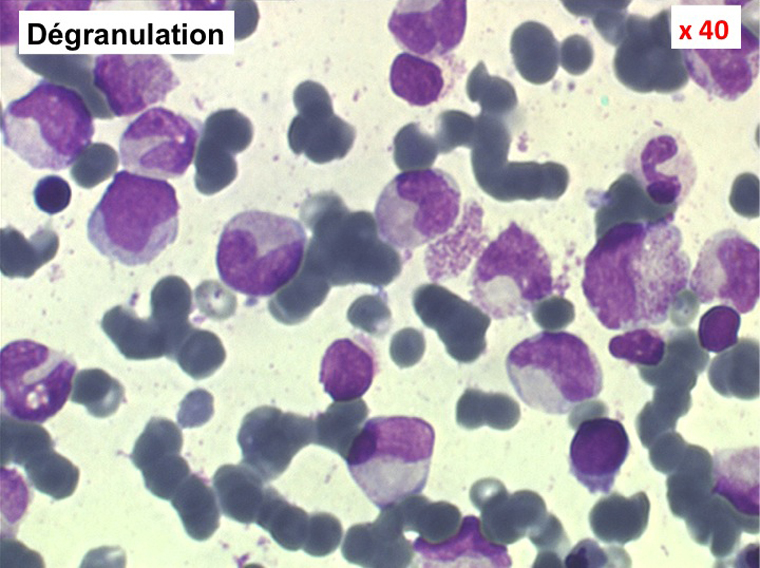
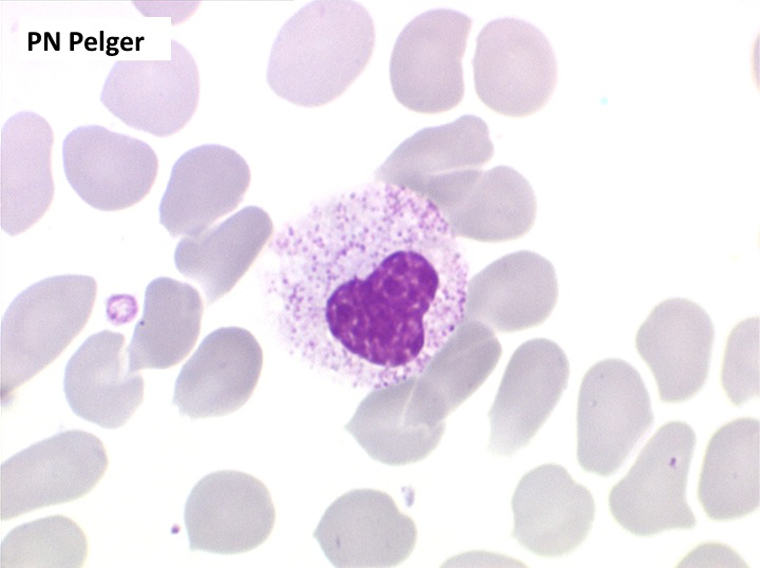
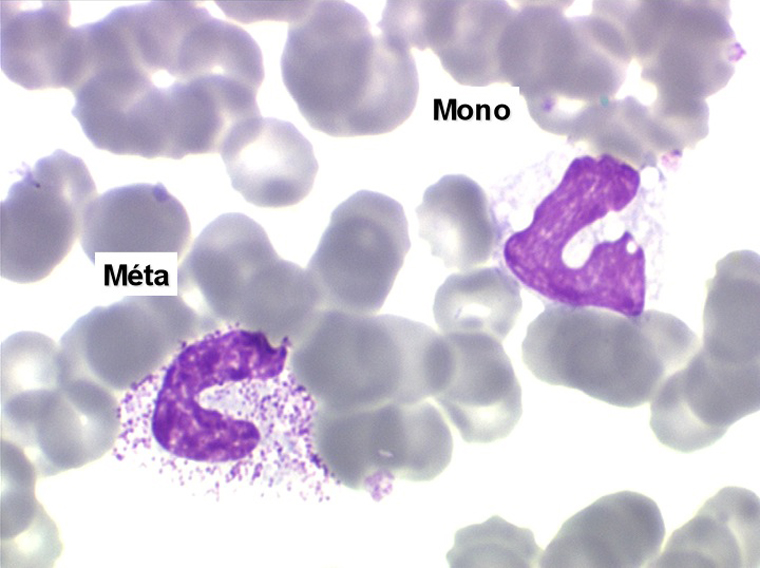
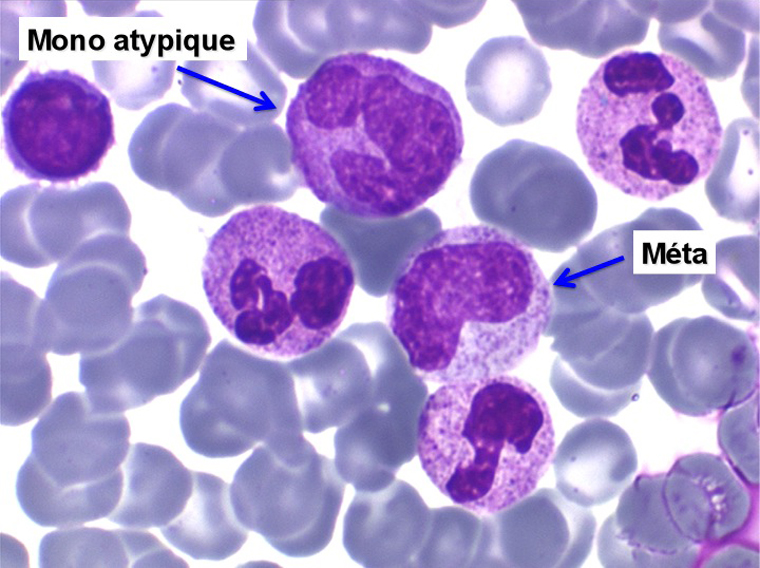
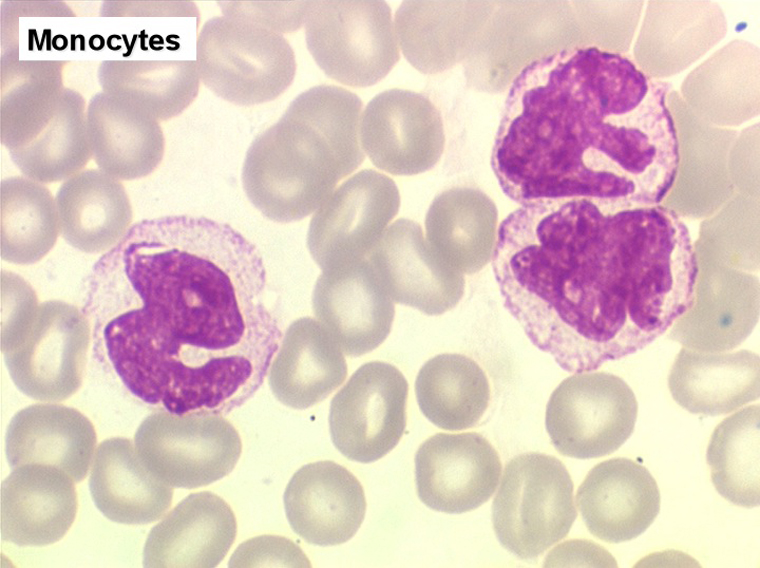
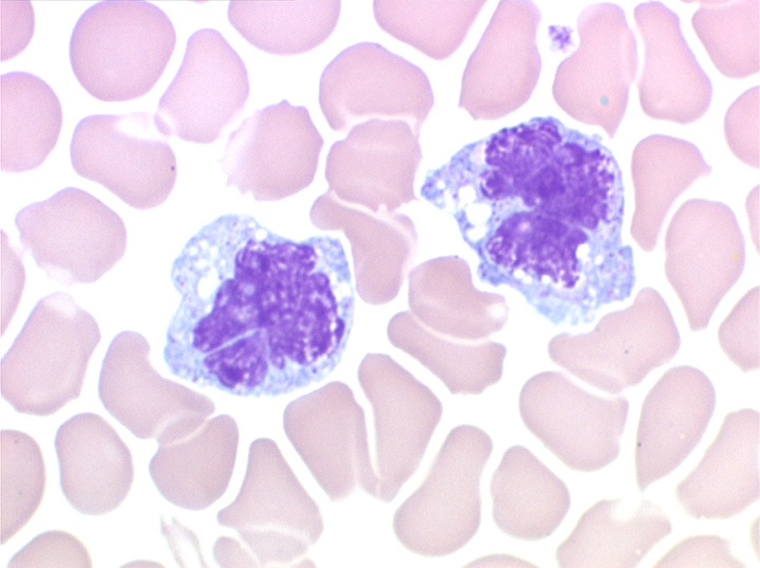
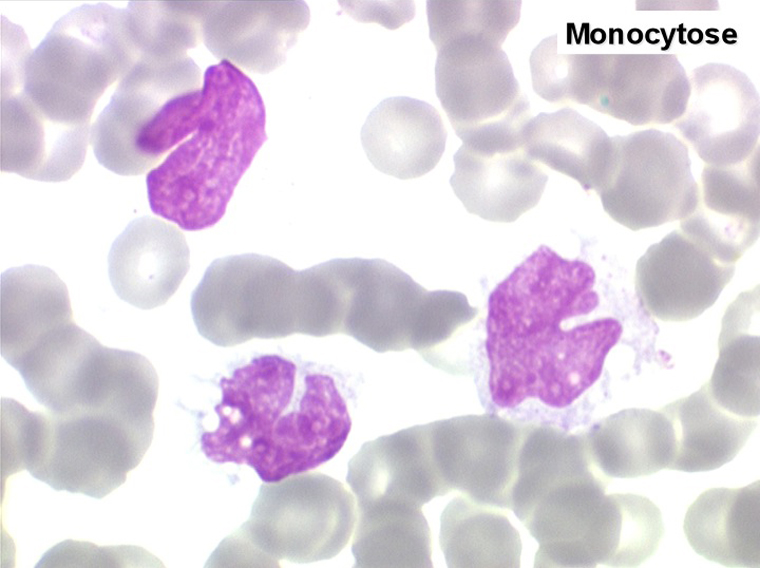
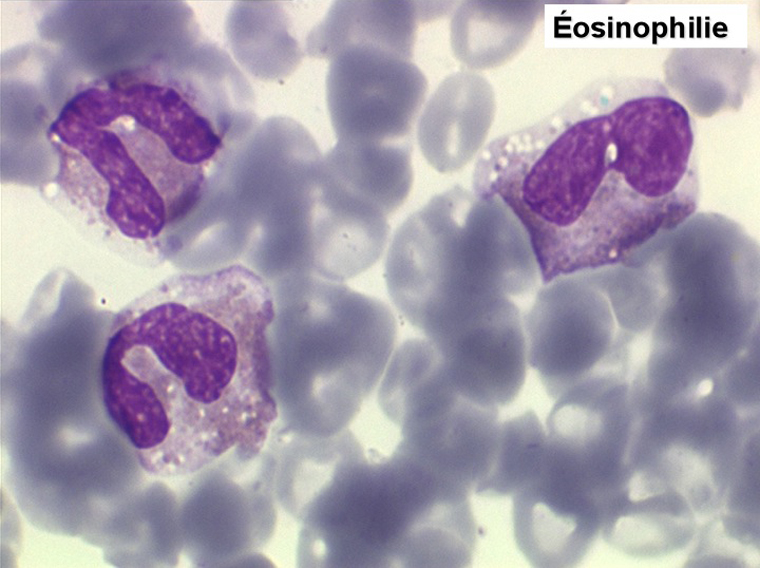
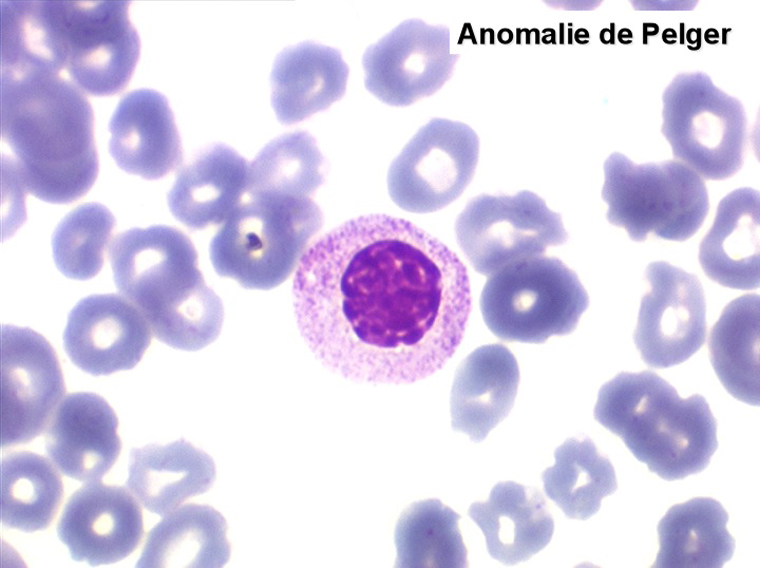
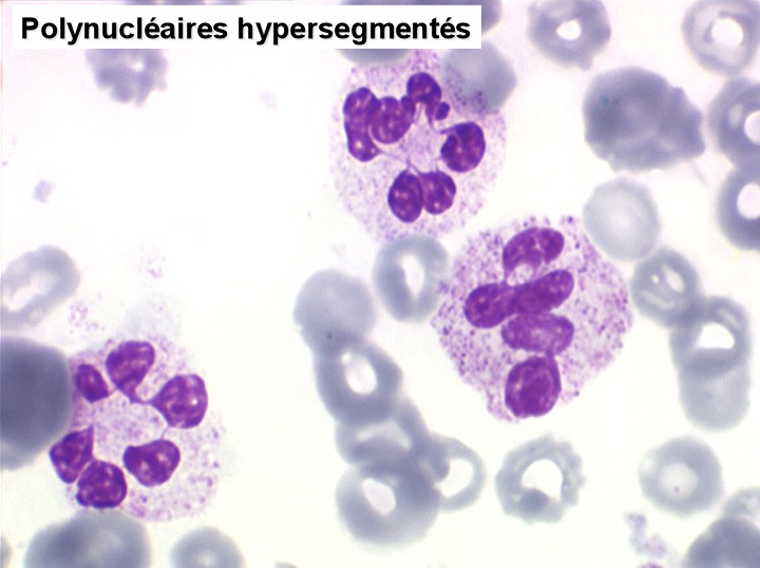
2. La leucopénie par neutropénie s'accompagne d'anomalies des polynucléaires : dégranulés, avec anomalie nucléaire de Pelger (neutrophiles non polylobés) ou au contraire hypersegmentés (dans la maladie de Biermer). Il y a souvent une discrète myélémie, une certaine éosinophilie et une monocytose.
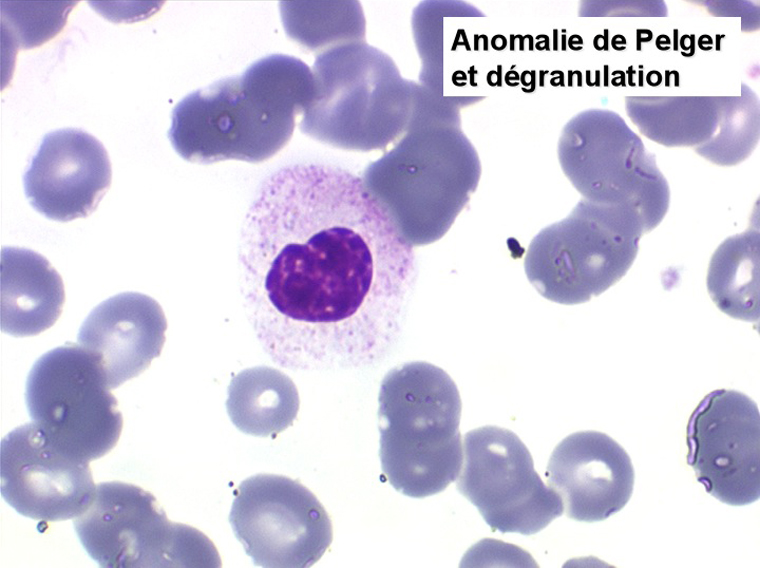
3. Les plaquettes peuvent ou non être diminuées, la présence de plaquettes géantes est fréquente et l'on peut trouver dans le sang des micro-mégacaryocytes ou des morceaux de cytoplasme de mégacaryocytes.
Les manifestations médullaires des myélodysplasies sont non moins variées
1. La moelle est riche, avec un déséquilibre entre les lignées en faveur des érythroblastes (moelle érythroblastique).
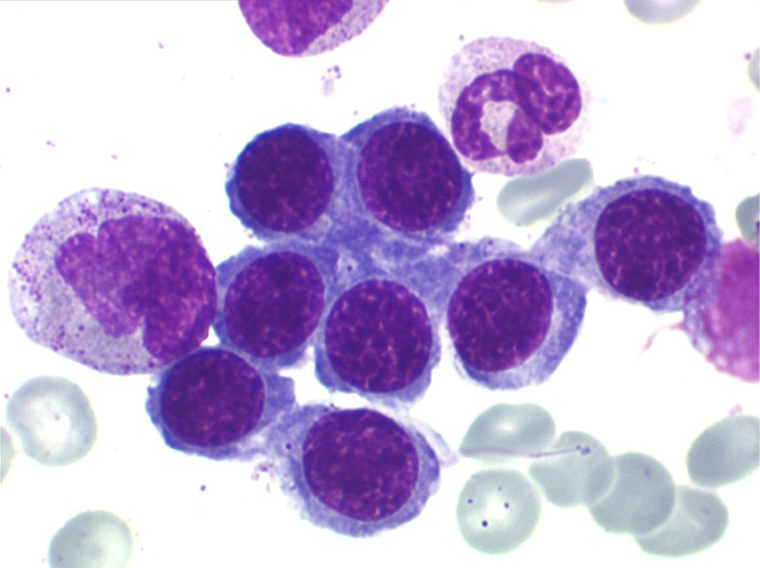
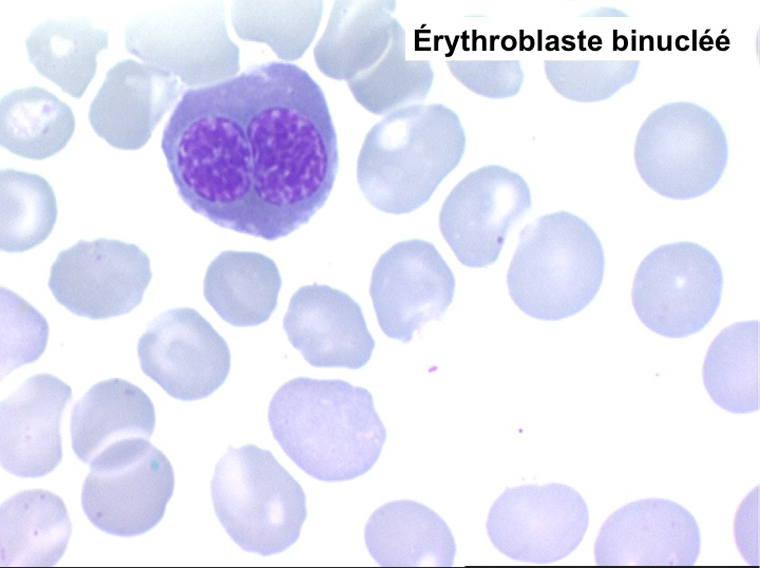
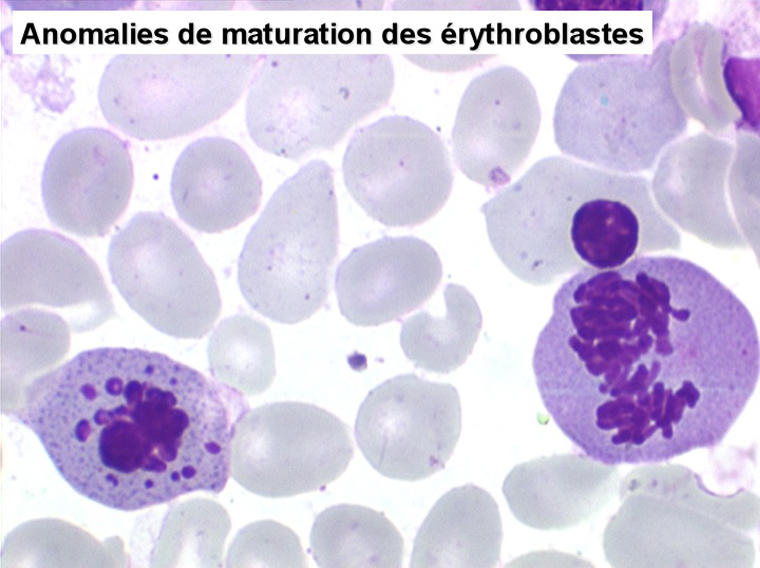
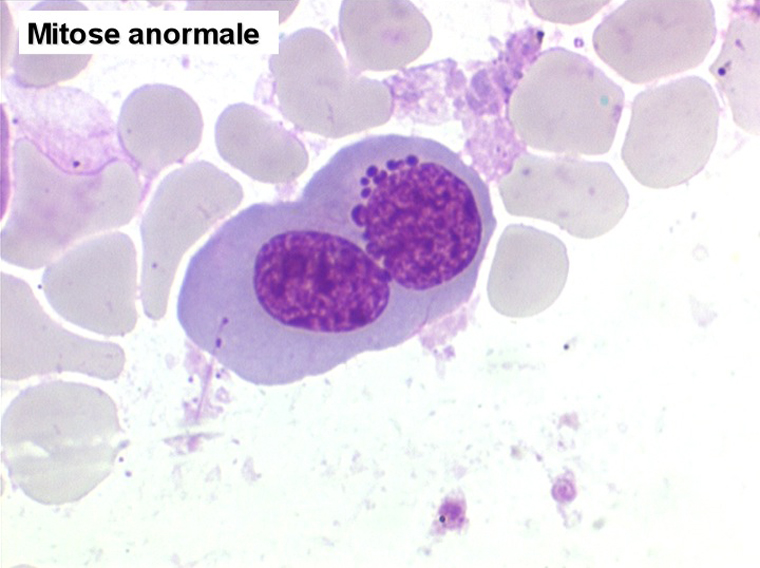
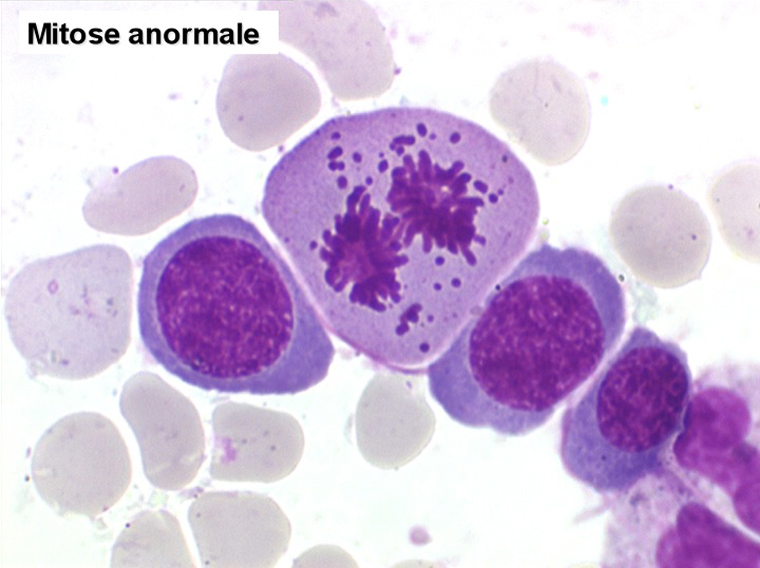
2. La dyshématopoïèse est manifeste au niveau de toutes les lignées.
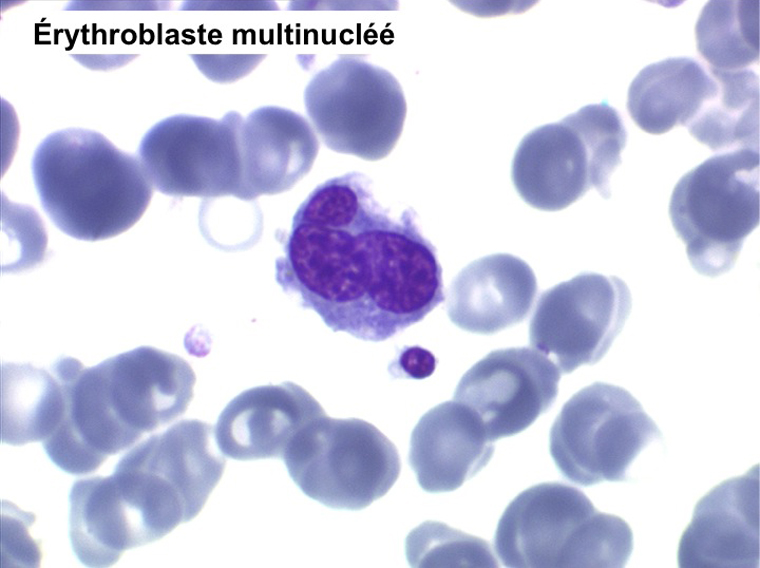
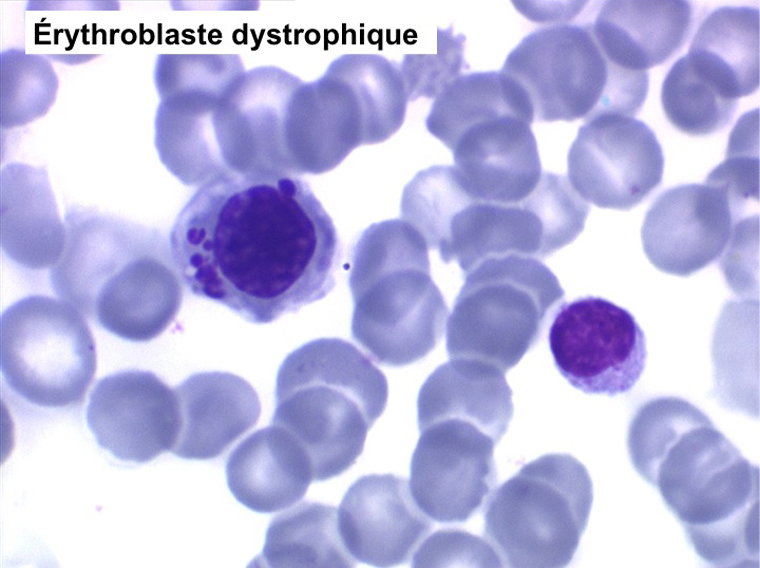
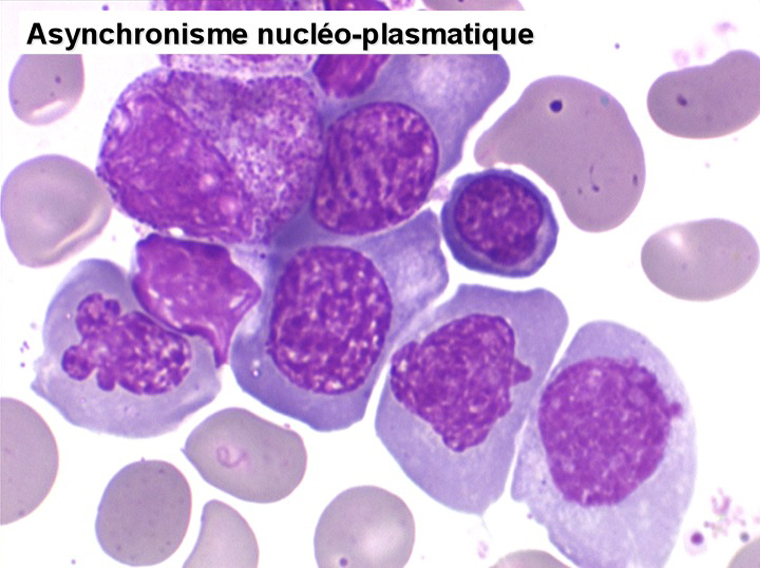
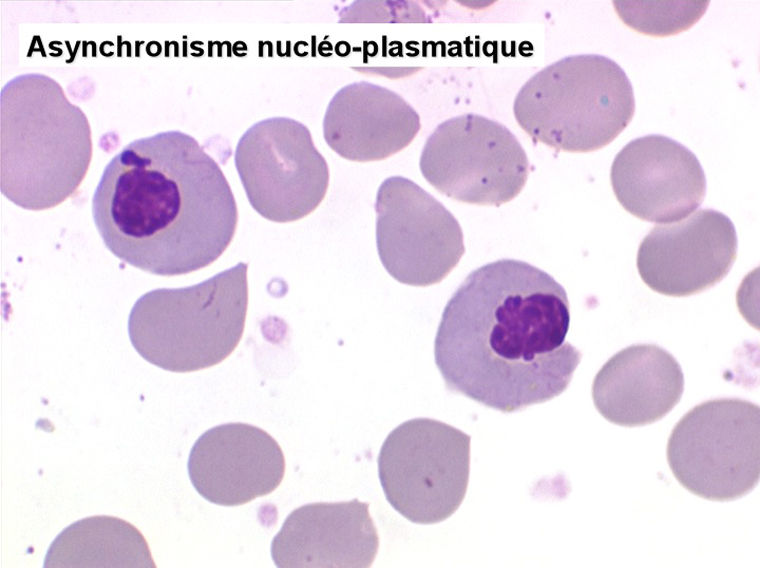
• La lignée rouge est généralement la plus touchée (dysérythropoïèse) avec une mégaloblastose, des érythroblastes plurinucléés, un net asynchronisme de maturation nucléo-plasmatique avec des noyaux tachetés en « peau de panthère », une tendance à la fragmentation anarchique du noyau dans les érythroblastes les plus mûrs, des vacuoles dans le cytoplasme.
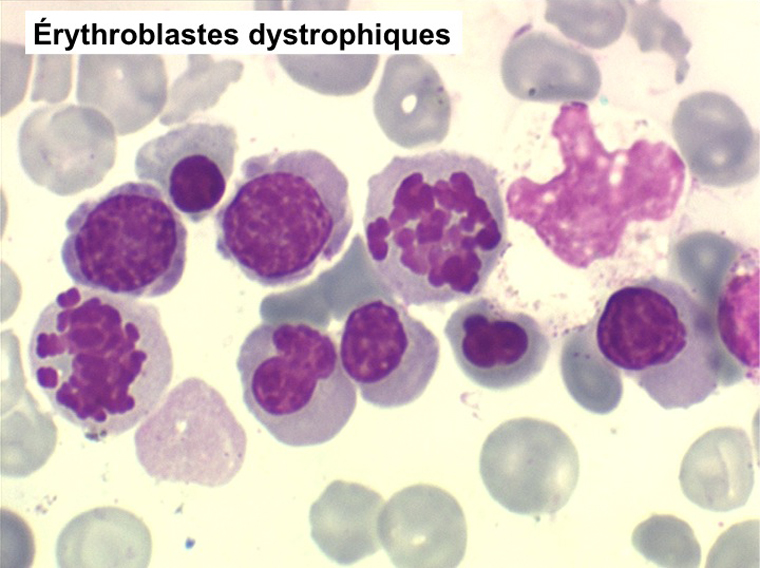
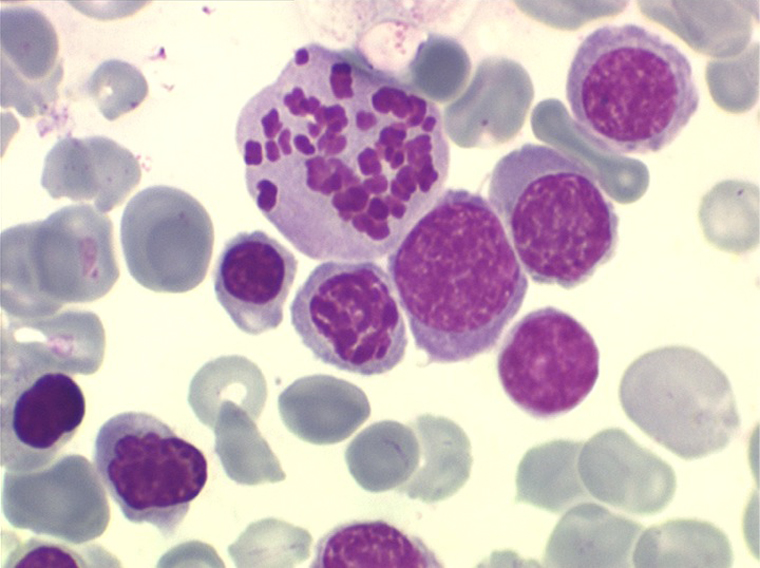
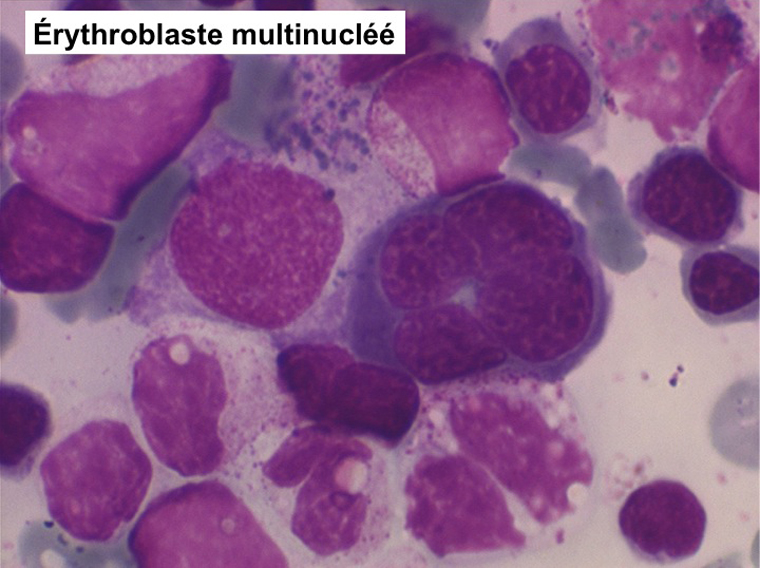
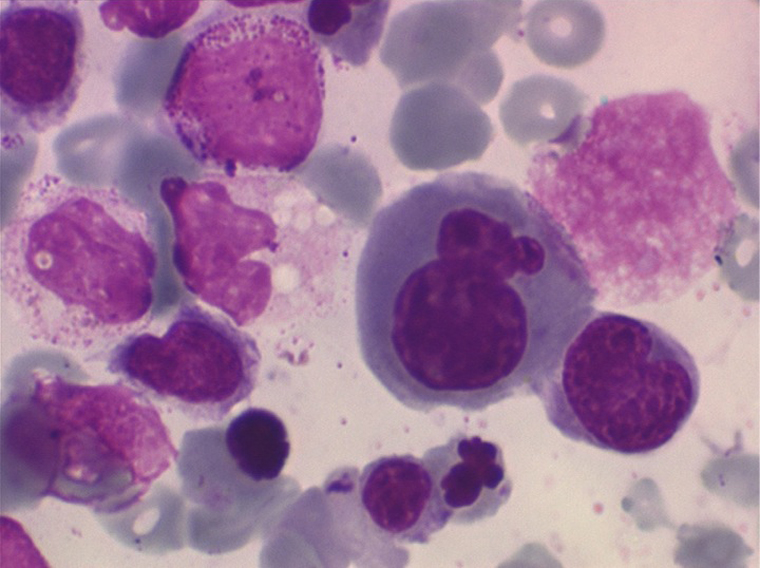
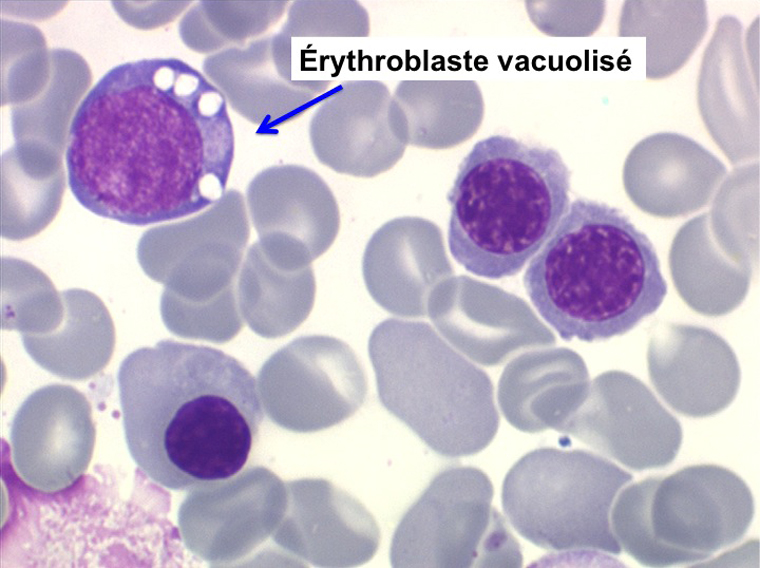
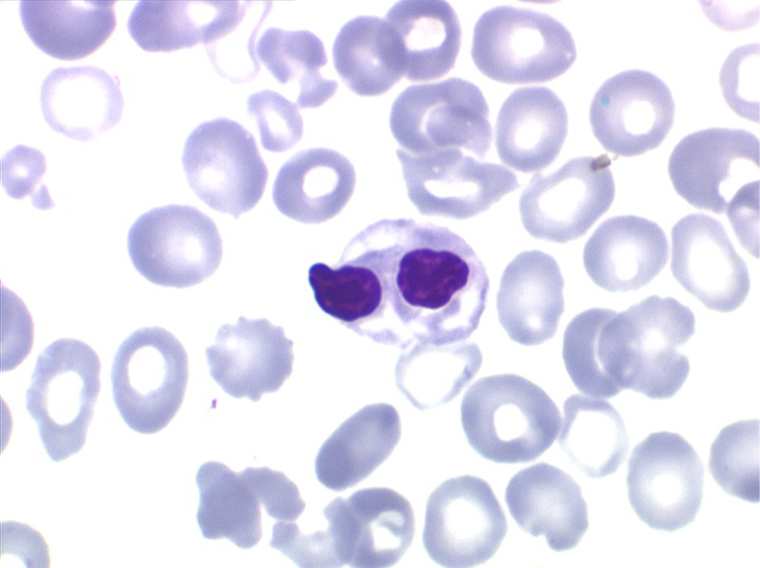
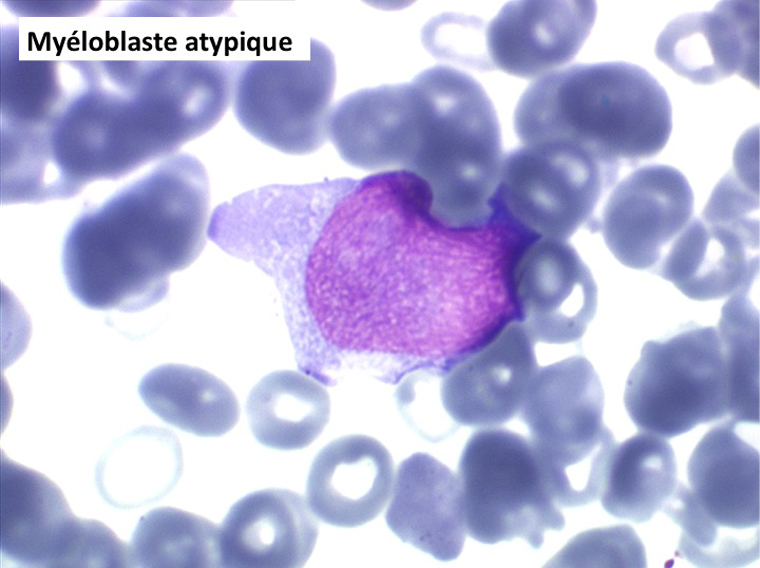
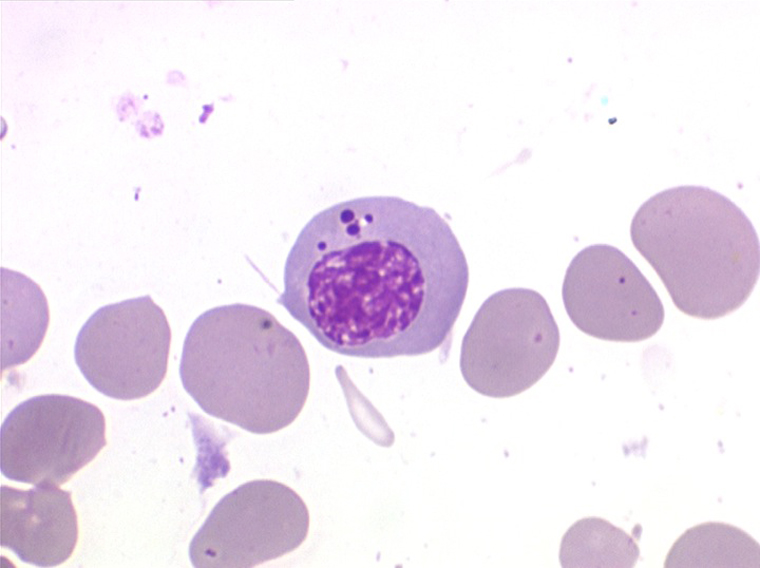
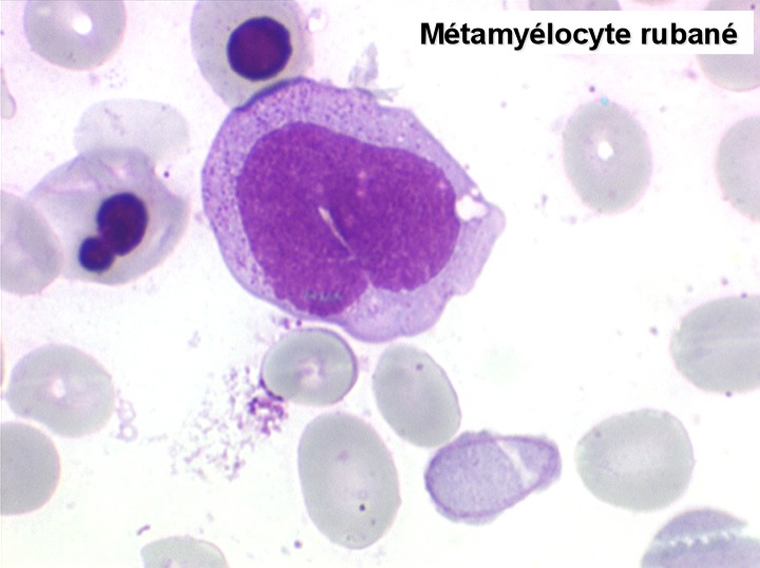
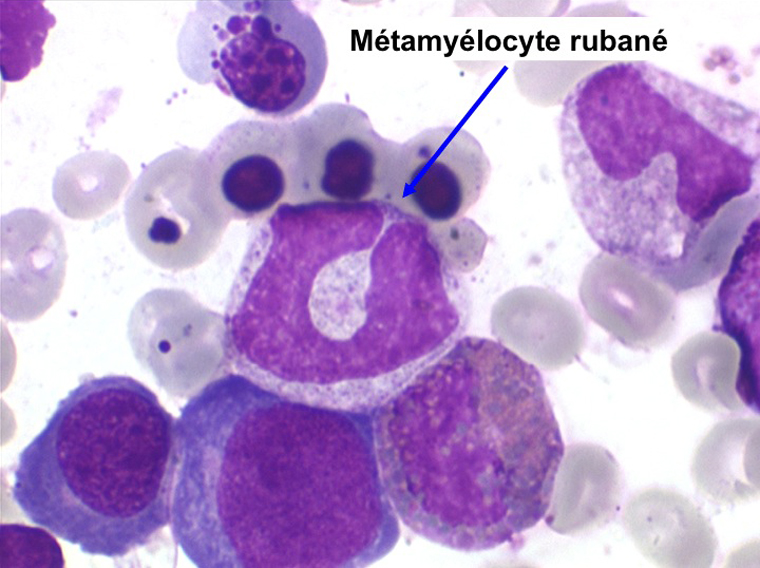
• La lignée myéloïde (dysgranulopoïèse) montre un excès de myéloblastes et de promyélocytes, les myélocytes et les métamyélocytes sont dégranulés et de ce fait de reconnaissance malaisée, pouvant être confondus avec des monocytes, d'autant que ceux-ci sont augmentés. Les métamyélocytes peuvent être de grande taille et présenter un noyau rubané. Il y a souvent une petite éosinophilie et un pourcentage accru de plasmocytes. Il faut compter avec soin les myéloblastes puisque la transformation en leucémie aiguë est définie par un taux de myéloblastes excédant 20%.
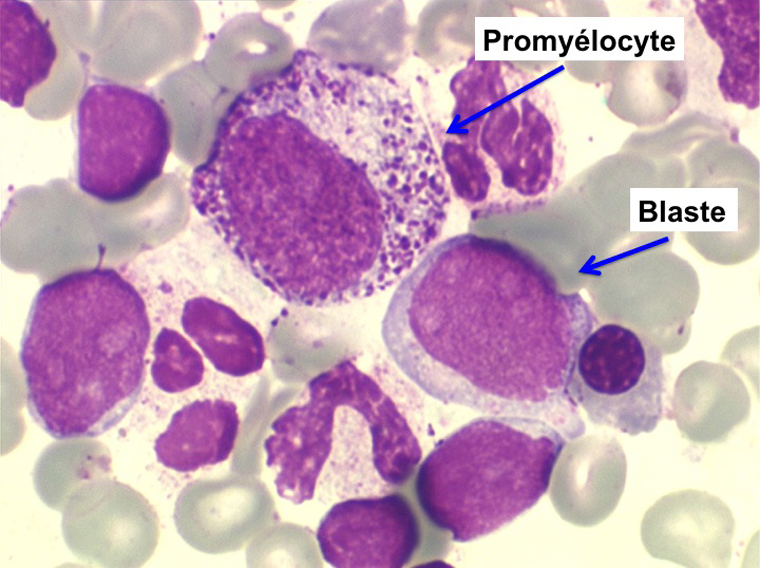
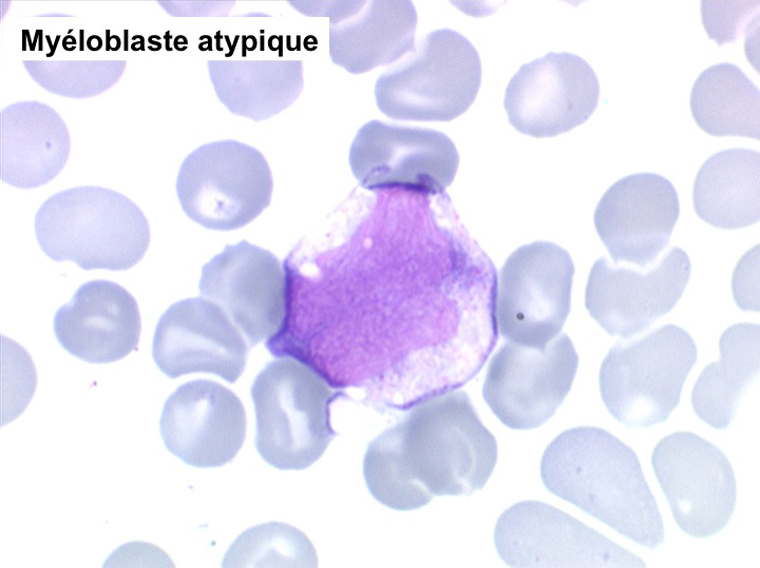
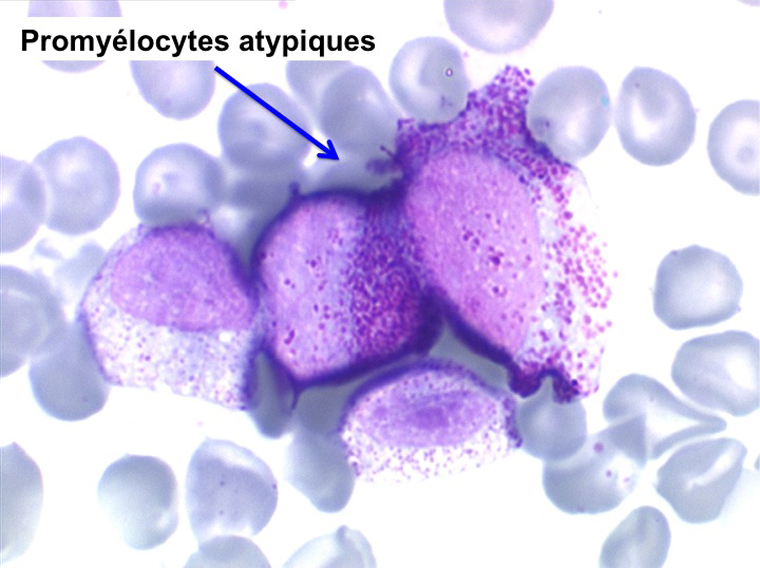
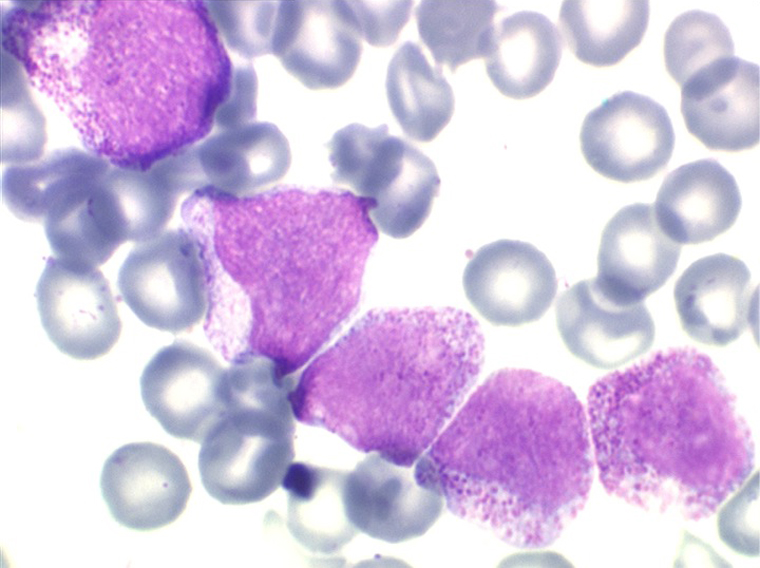
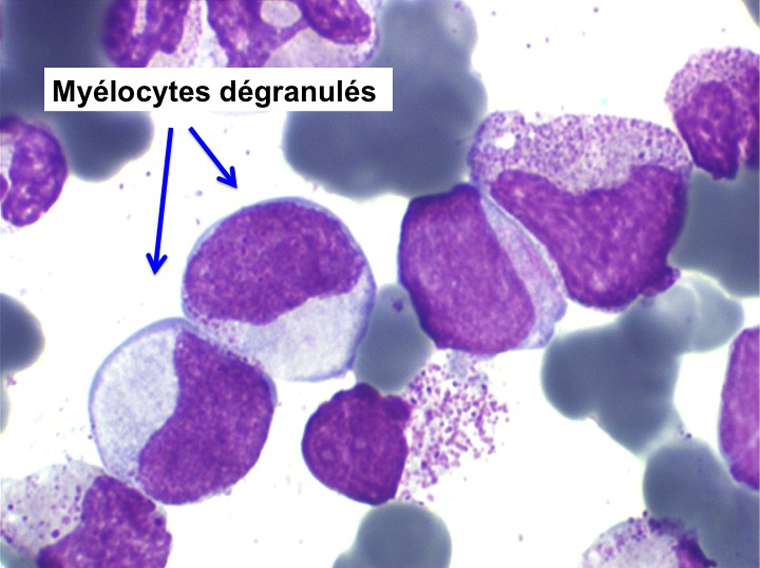
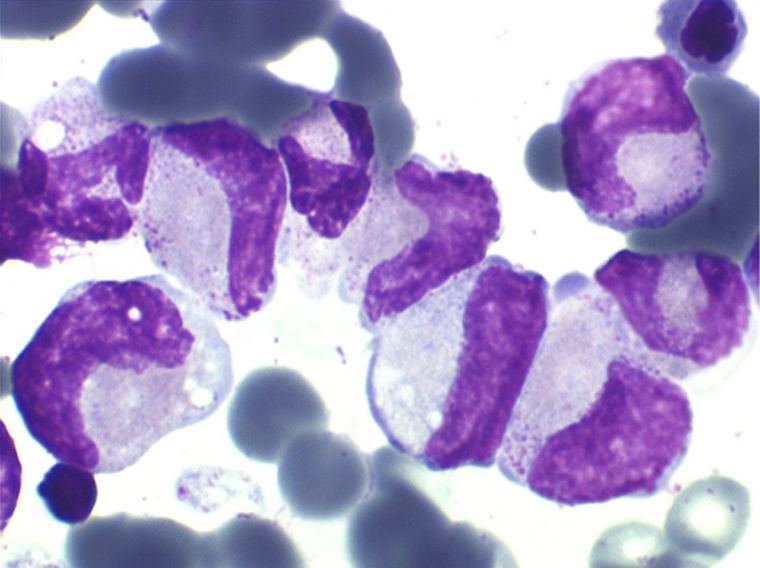
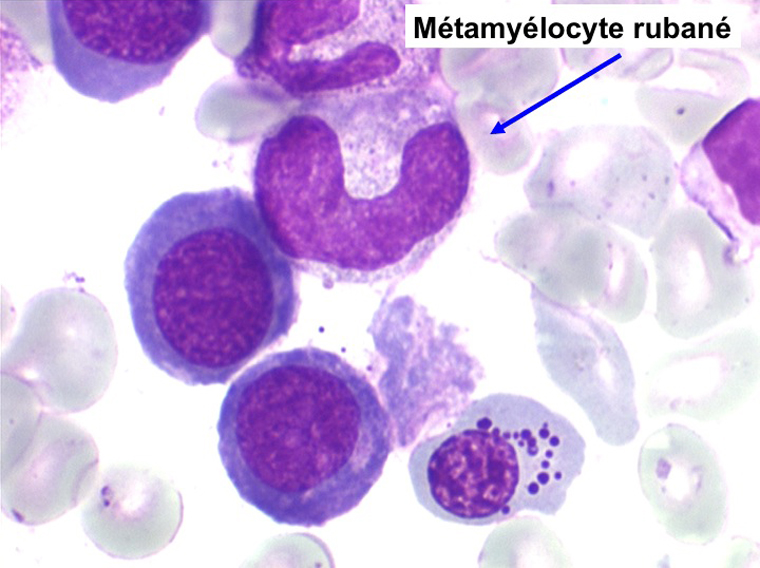
• La lignée mégacaryo-plaquettaire est elle aussi touchée avec des mégacaryocytes anormaux, de petite taille.
2 – Les myélodysplasies primitives
Les myélodysplasies primitives sont des maladies des cellules souches pluripotentes, monoclonales, par lésions géniques acquises, idiopathiques dans l'état actuel de nos connaissances. Cette atteinte est multilignée et responsable d'une hématopoïèse inefficace qui entraîne une cytopénie sanguine périphérique portant sur tout ou partie des éléments sanguins dont le premier temps est habituellement, mais pas toujours, une anémie, et des dystrophies morphologiques plus ou moins importantes des éléments médullaires et sanguins. Le tableau hématologique abouti est le plus souvent celui d'une « pancytopénie à moelle riche ». Comme pour les syndromes myéloprolifératifs l'unicité de ces syndromes est dans la fréquence de survenue, à la phase terminale, d'une leucémie aiguë secondaire, habituellement réfractaire au traitement.
Les examens biologiques nécessaires au diagnostic de myélodysplasie primitive comportent, outre l'étude quantitative et morphologique du sang et de la moelle osseuse, la cytochimie des éléments sanguins et médullaires (notamment myéloperoxydases et coloration de Perls pour compter et quantifier les sidéroblastes), la culture des progéniteurs des lignées médullaires (notamment des CFU-GM dont la pousse est anormale et le nombre des colonies diminué et de petite taille), l'étude cytogénétique des cellules médullaires à la recherche d'éventuelles anomalies chromosomiques, les dosages biochimiques des lysozymes sanguins et urinaires, de la LDH et du métabolisme ferrique. Les syndromes myélodysplasiques primitifs peuvent se scinder en 4 entités anatomo-cliniques différentes :
• l'anémie réfractaire (AF),
• l'anémie réfractaire avec excès de blastes (AREB),
• l'anémie sidéroblastique idiopathique acquise (ASIA),
• la leucémie myélo-monocytaire chronique (LMMC).
(1) – L'anémie réfractaire (AF)
Comme son nom l'indique il s'agit d'une anémie ressemblant à l'anémie mégaloblastique de Biermer mais réfractaire au traitement par la vitamine B12. En effet elle n'est pas due à une avitaminose mais est le premier stade d'une myélodysplasie primitive. L'anémie est macrocytaire, volontiers hypochrome. Les anomalies des polynucléaires et des plaquettes sont au début peu importantes mais apparaissent progressivement et, par définition, il n'y a pas ou peu de myéloblastes circulants (moins de 1%).
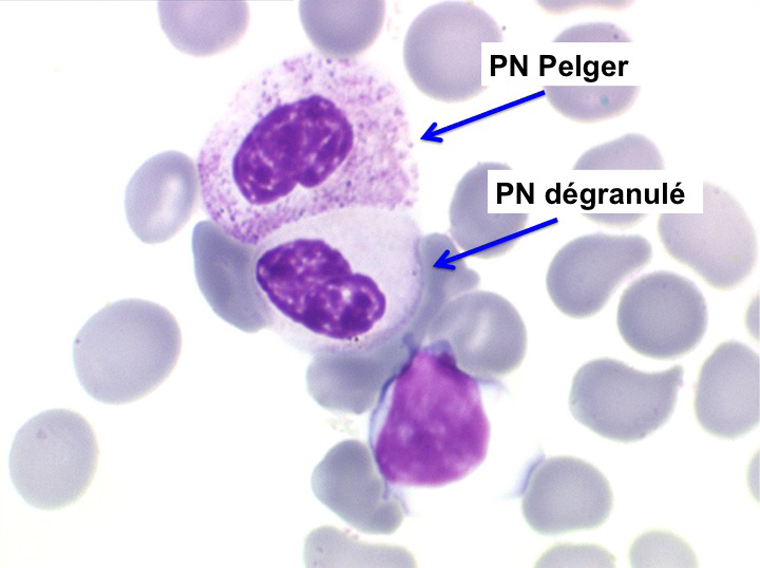
La moelle, riche, contient moins de 5% de blastes. L'évolution de cette anémie est chronique, elle s'accentue progressivement nécessitant habituellement des transfusions sanguines, le tableau peut se compléter avec apparition de blastes (tableau d'AREB), la transformation en leucémie aiguë n'est pas rare (10% à 15% des cas).
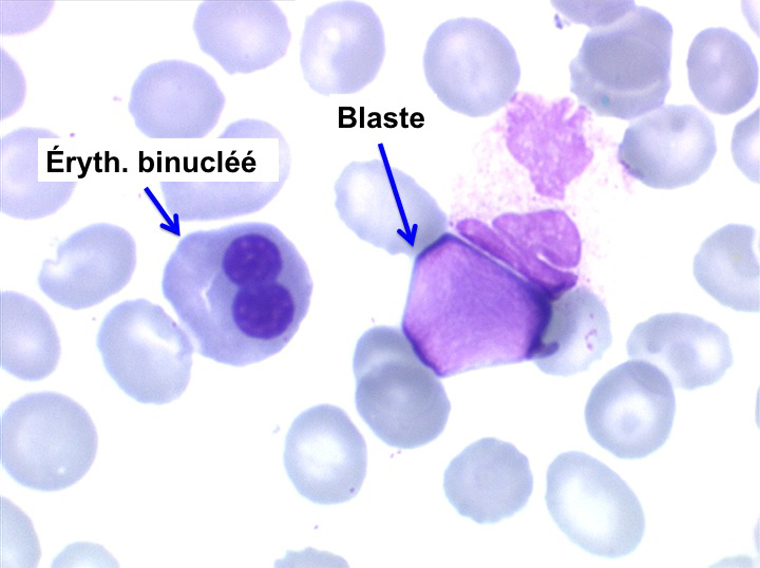
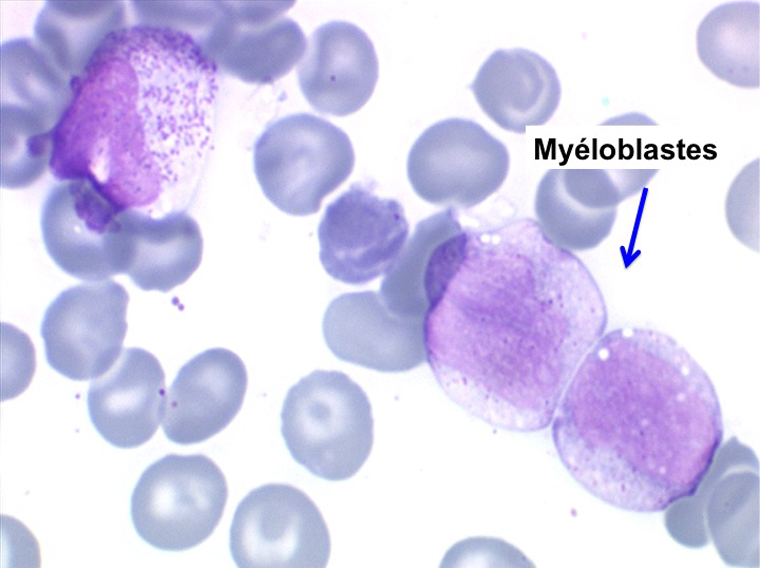
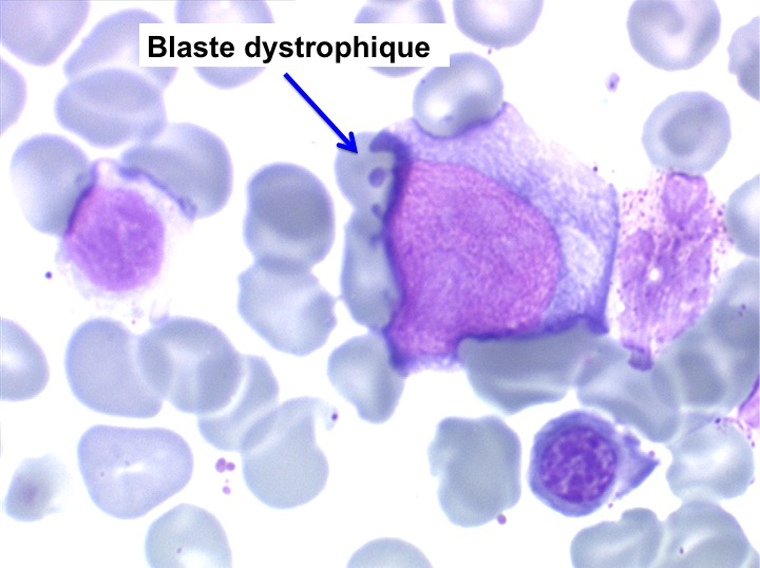
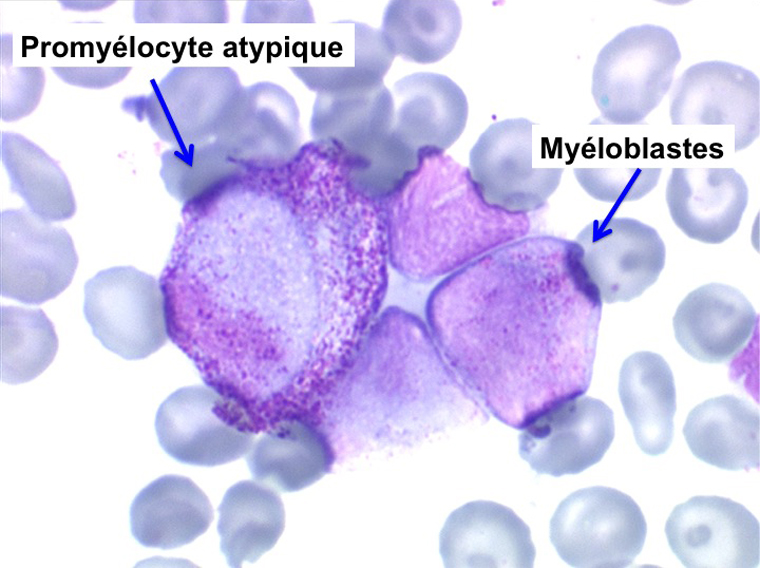
L'anémie n'est pas toujours le premier symptôme d'une myélodysplasie, plus rarement le début est une leuco-neutropénie isolée, chronique, d'origine centrale (test de démargination négatif) qui ne se complétera par une anémie réfractaire que de nombreuses années plus tard. Plus rarement encore le début peut être une thrombopénie longtemps isolée.
Une forme particulière d'anémie réfractaire s'accompagne d'une anomalie cytogénétique spécifique, le syndrome 5q-, il atteint des femmes âgées (2/3 des cas) et se caractérise, outre l'anémie macrocytaire, par une thrombocytémie importante (> 1 million/µl) avec dans la moelle des mégacaryocytes anormaux, non polylobés.
(2) – L'anémie réfractaire avec excès de blastes (AREB)
Comme son nom l'indique il s'agit d'une anémie ayant les mêmes caractéristiques que la forme précédemment décrite mais avec, dans la sang et dans la moelle, présence de précurseurs de granuleux (myéloblastes et promyélocytes). Ce taux ne doit cependant pas dépasser 5% dans le sang et 20% dans la moelle, au-delà en effet on ne peut plus parler de myélodysplasie mais de véritable leucémie aiguë myéloblastique avec myélodysplasie, forme à présent isolée dans la classification OMS des leucémies aiguës. Quoiqu'il en soit le devenir le plus fréquent (30% à 50% des cas) des AREB est de se transformer, après un temps plus ou moins long d'évolution de l'anémie chronique, en leucémie aiguë.

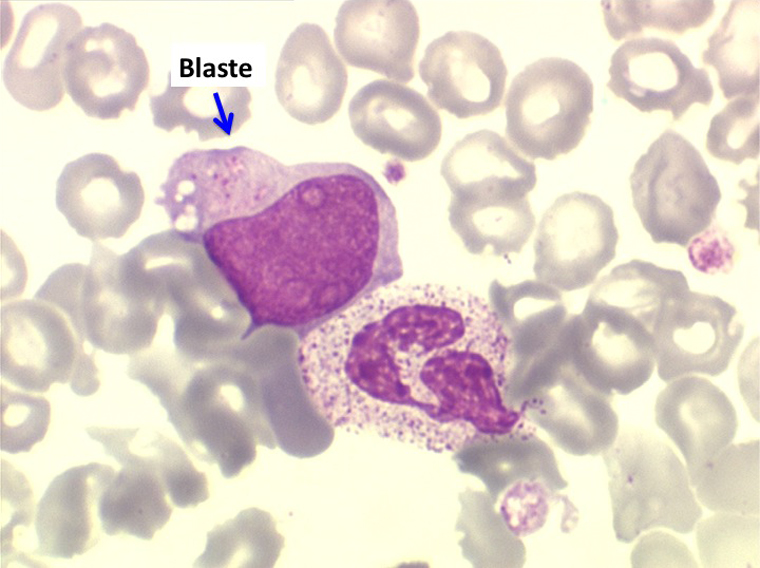
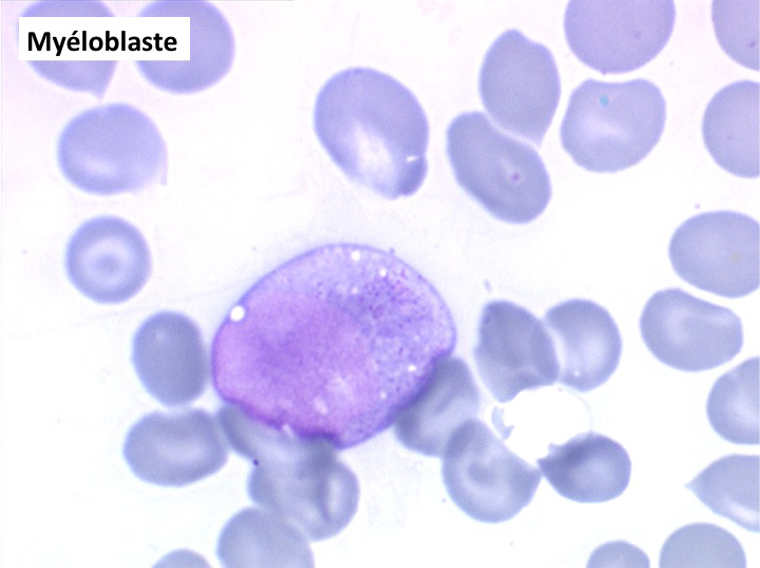
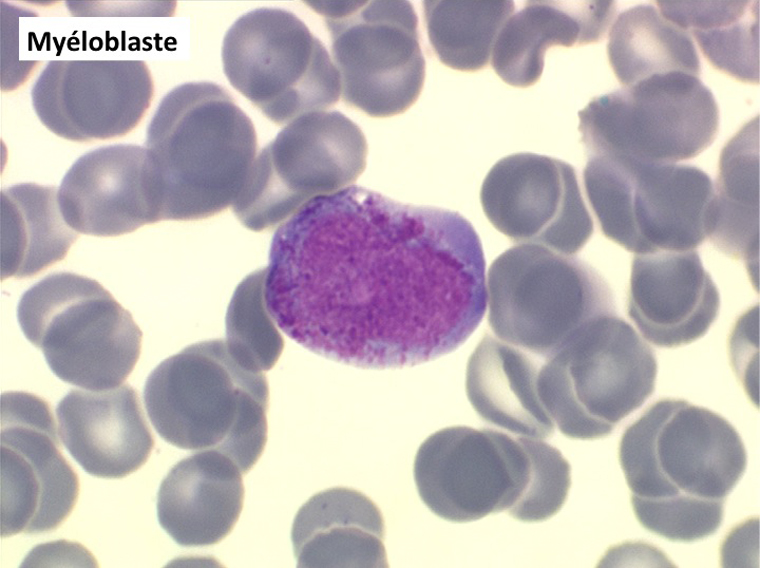
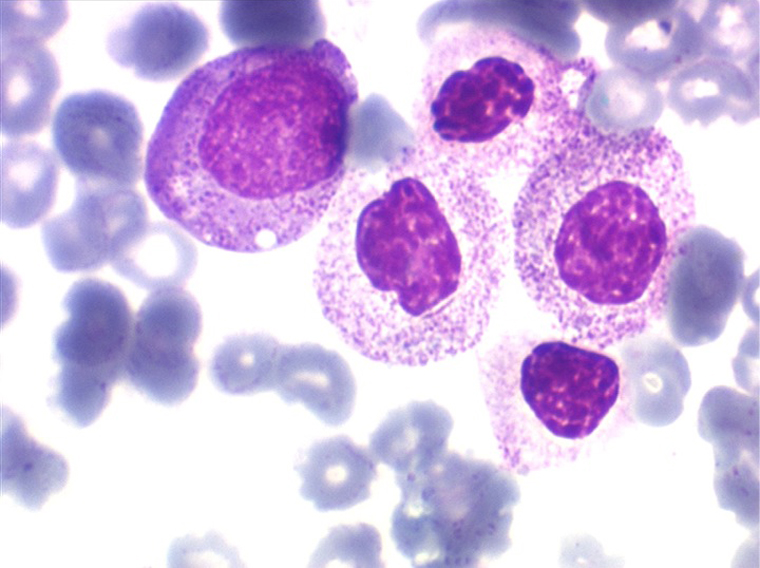
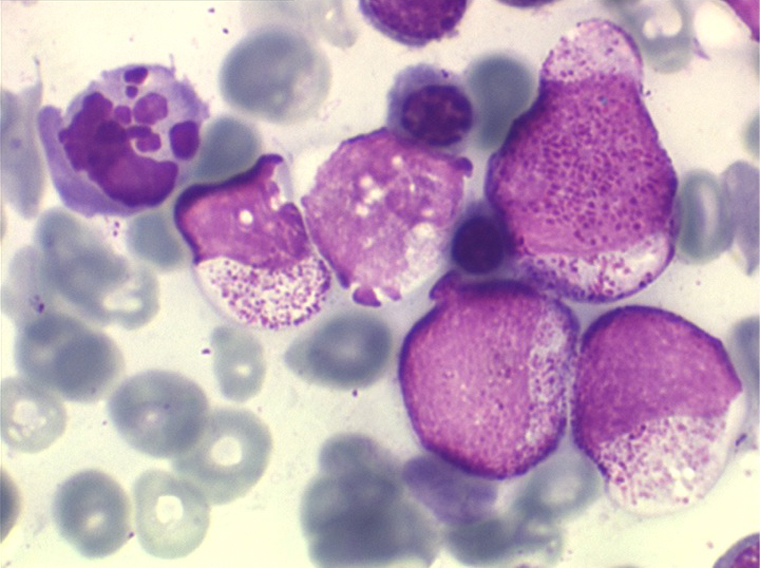
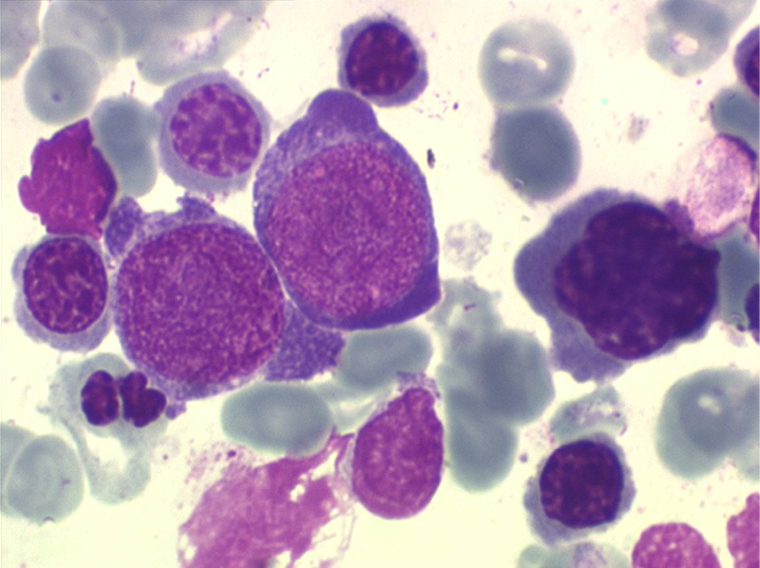
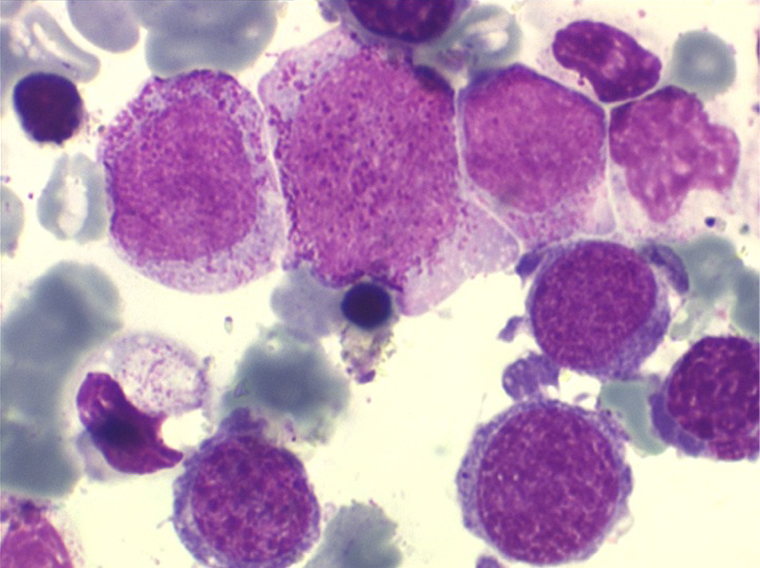
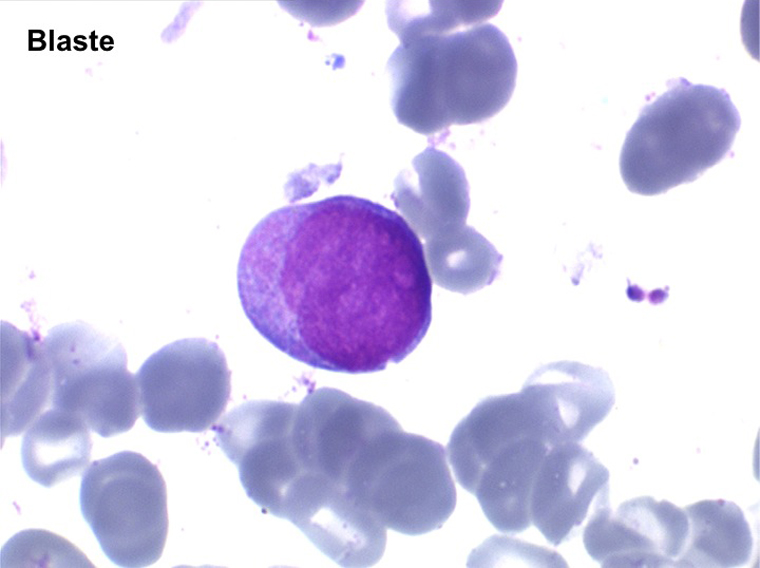
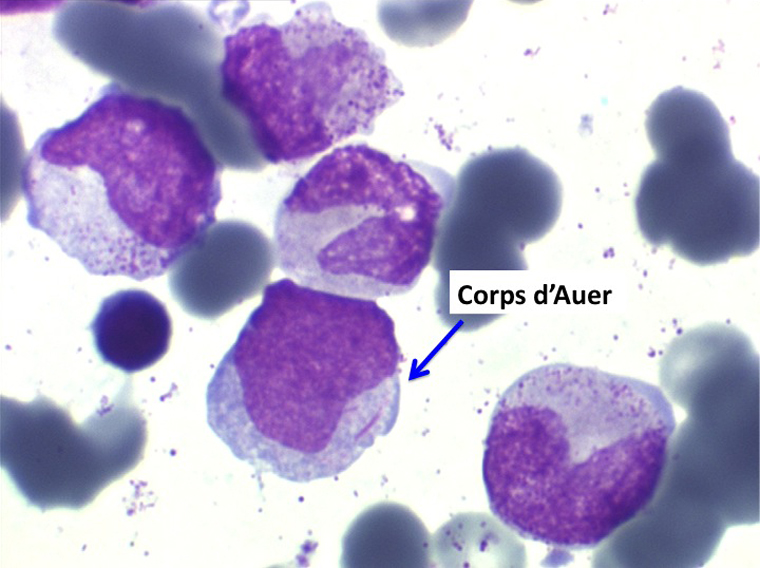
(3) – L'anémie sidéroblastique idiopathique acquise (ASIA)
L'anémie sidéroblastique idiopathique acquise revêt le tableau d'une anémie réfractaire mais est caractérisée par la présence dans la moelle d'un grand nombre d'érythroblastes plus ou moins dystrophiques dont la plupart sont des sidéroblastes, fortement chargés en fer, dont certains (>15%) ont une répartition des grains de fer en couronne autour du noyau. Cette sidéroblastose médullaire est le reflet d'une anomalie de synthèse de l'hémoglobine dont la traduction sanguine est une hypochromie et une élévation du fer sérique et de la ferritine (hypersidérémie) traduisant une surcharge des stocks de fer de l'organisme. L'anémie a les mêmes caractères que ceux de l'anémie réfractaire et, tout comme elle, s'il peut y avoir présence d'un excès de blastes dans le sang et dans la moelle, leur pourcentage n'excède pas 1% dans le sang et 5% dans la moelle. Le plus souvent le taux de plaquettes est normal, il peut même exister une thrombocytose, la moelle est riche en mégacaryocytes.
Comme pour les autres myélodysplasies, l'ASIA peut se transformer après quelques années d'évolution en leucémie aiguë. Mais le devenir habituel de l'ASIA est autre, l'anémie s'accentuant progressivement des transfusions sanguines sont nécessaires qui aggravent la surcharge en fer, l'hémosidérose progresse, responsable d'une hépato-splénomégalie et surtout d'une cardiomégalie avec insuffisance cardiaque. C'est celle-ci qui est la cause habituelle du décès des ASIA.
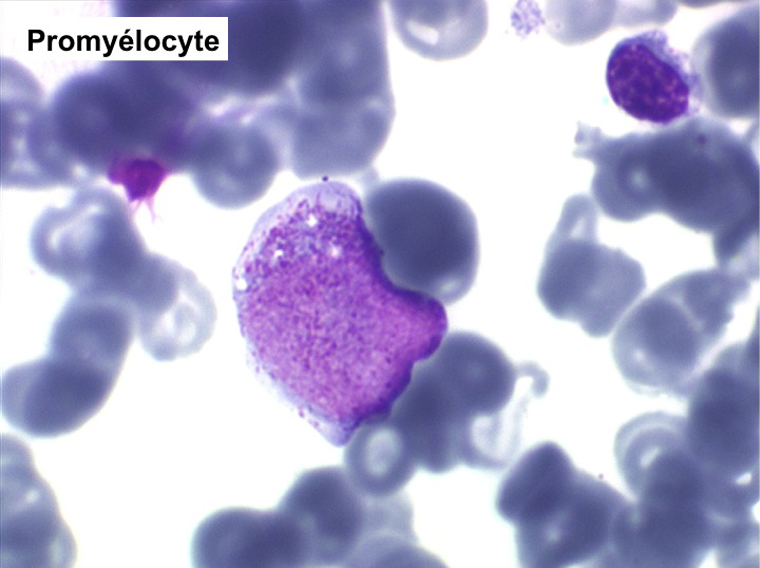
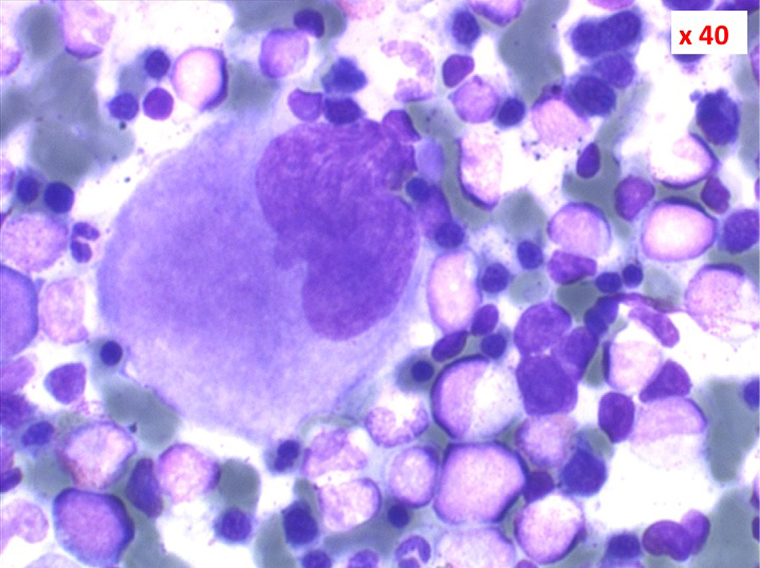
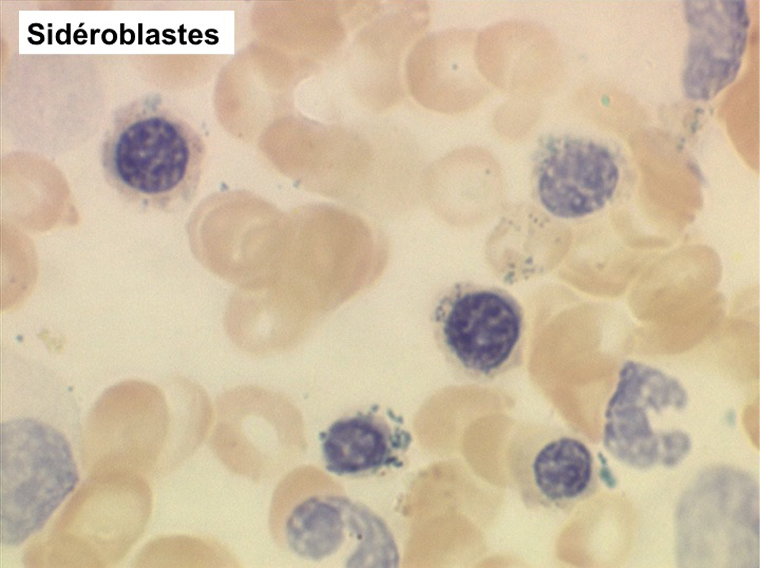
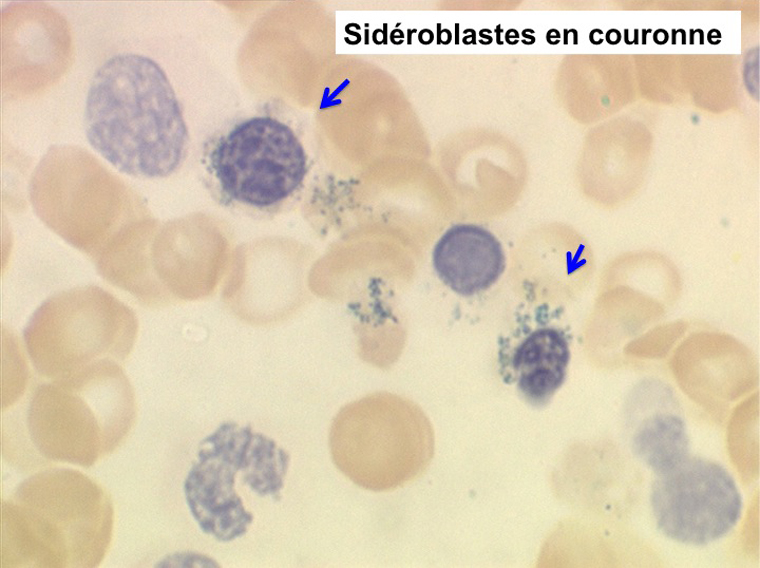
(4) – La leucémie myélo-monocytaire chronique (LMMC)
C'est une entité tout à fait à part dans le cadre des myélodysplasies. Sa caractéristique principale, à côté d'une anémie chronique de type réfractaire, est de comporter une importante monocytose sanguine, d'où le nom donné à cette maladie. Cette monocytose est > 1.000/µl, faite de monocytes typiques, habituellement peu dystrophiques. Le taux de blastes circulants, s'il y en a, est très faible. On retrouve la monocytose dans la moelle, mais il est souvent difficile de distinguer les monocytes des myélocytes et métamyélocytes car ces derniers sont dégranulés ou ont des granulations poussiéreuses identiques à celles des monocytes. Le taux de myéloblastes et promyélocytes médullaires n'est habituellement pas très augmenté, mais on retrouve des monoblastes et des promonocytes, le taux total des cellules jeunes ne doit cependant pas excéder 20%. Le diagnostic sera étayé par le dosage des lysozymes sanguins et urinaires dont l'augmentation est le reflet du turn-over important des monocytes. L'évolution est d'une grande chronicité et les traitements myélofreinateurs ne sont que rarement nécessaires et toujours risqués. L'évolution terminale en leucémie aiguë n'est pas rare et s'observe dans 30 à 40% des cas.

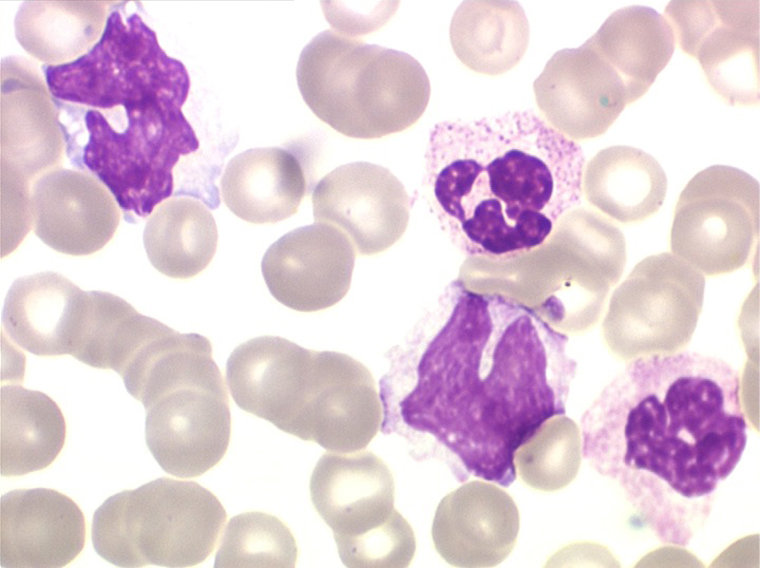
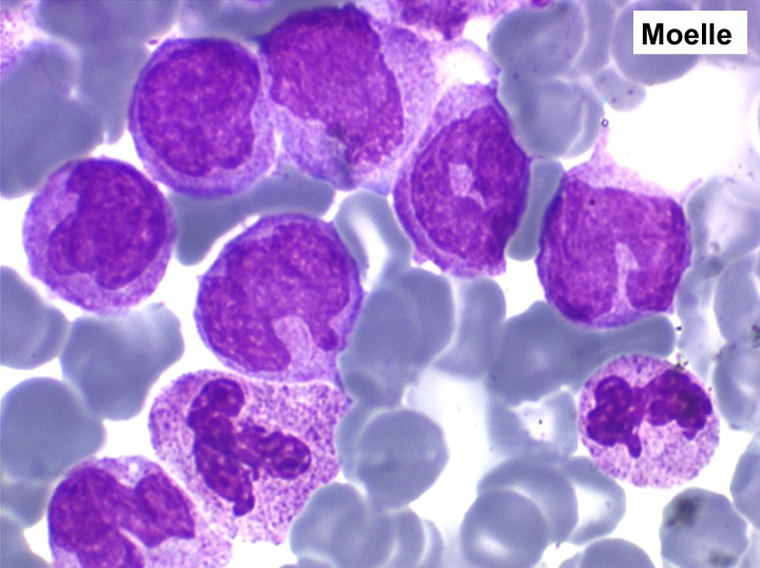
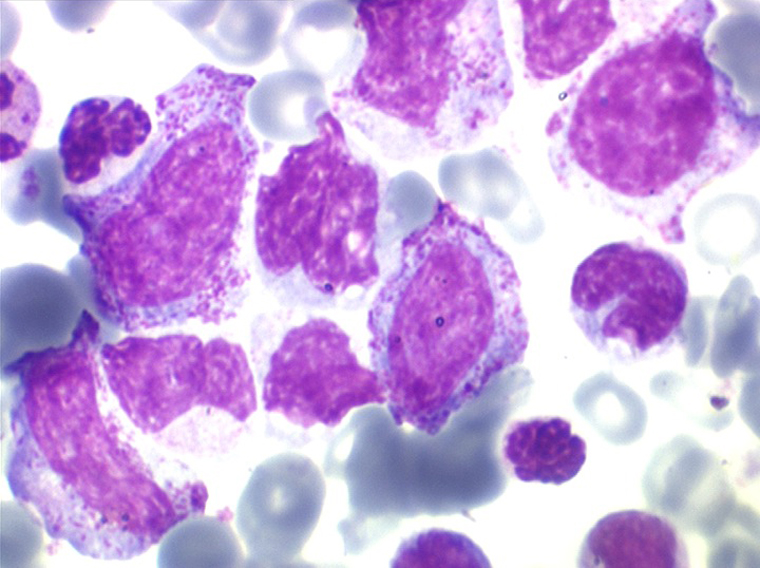
3 – Les myélodysplasies secondaires
Les myélodysplasies secondaires sont les plus fréquentes.
(1) – Une des causes de cette fréquence est que ce type d'altération médullaire apparaît et s'accentue avec l'âge, constituant une manière quasi physiologique du vieillissement de la moelle osseuse, on ne peut donc que voir augmenter les syndromes myélodysplasiques au fur et à mesure du vieillissement de la population.
(2) – Une autre cause fréquente de syndrome myélodysplasique est la chimiothérapie anticancéreuse, surtout les polychimiothérapies. Chaque cycle de chimiothérapie entraine une aplasie médullaire transitoire dont l'intensité dépend des produits utilisés et de leurs doses mais aussi du type de cancer (avec localisation médullaire ou non). Cette aplasie transitoire met de plus en plus de temps pour se réparer au fur et à mesure du déroulement de la chimiothérapie, mais surtout des clones médullaires anormaux apparaissent donnant un tableau de myélodysplasie progressive. Celle-ci régresse dans les mois qui suivent l'arrêt du traitement, mais dans quelques cas la myélodysplasie persiste et progresse, pouvant aboutir à une leucémie secondaire. Le tableau cytologique de la myélodysplasie chimiothérapique n'a rien de particulier et est semblable à celui d'une anémie réfractaire. On peut noter cependant :
• La constance et la précocité de la macrocytose qui très souvent persiste après l'arrêt du traitement.
• L'apparition progressive, d'un cycle de chimiothérapie à l'autre, des stigmates sanguins de myélodysplasie, notamment la tendance à la dégranulation des polynucléaires et l'apparition d'anomalies de Pelger.
• L'existence d'une « vague » de monocytose sanguine, parfois importante et de morphologie dystrophique, à la phase de réparation de l'aplasie, quelques jours avant la réapparition des polynucléaires.
• Une éosinophilie, surtout avec certains médicaments (Endoxan).
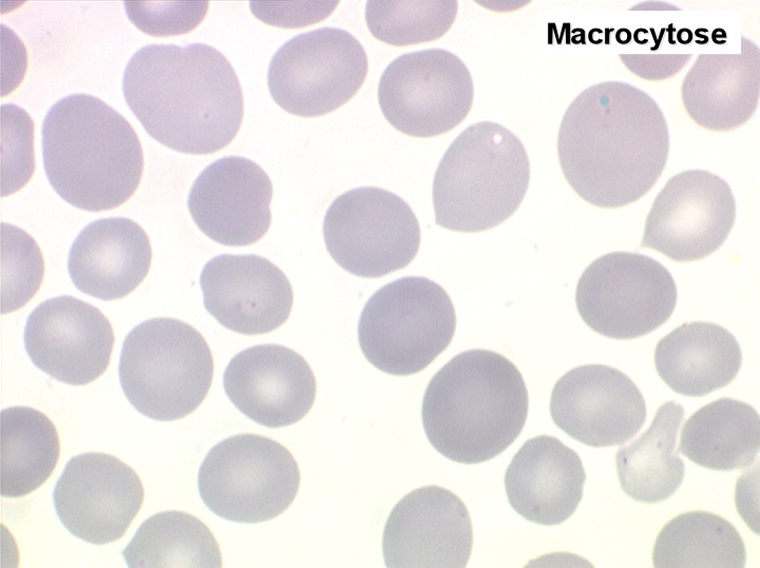
(3) – On voit moins de myélodysplasies dues à des avitaminoses : anémie mégaloblastique de Biermer par avitaminose B12 ou anémie macrocytaire par carence en acide folique. La maladie de Biermer mérite cependant qu'on s'y arrête car elle réalise le tableau hématologique le plus pur de myélodysplasie, notamment dans les dystrophies de la moelle osseuse. Il s'agit en fait d'une maladie de l'estomac (achylie gastrique) responsable d'une absence de sécrétion du « facteur Intrinsèque » indispensable à l'assimilation de la vitamine B12. La carence en vitamine B12 qui s'ensuit entraine des troubles multiples, notamment sanguins et neurologiques.
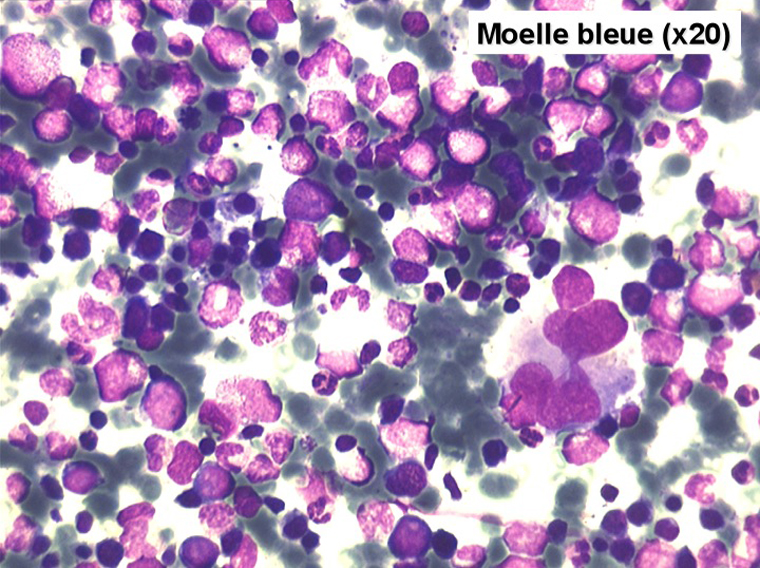
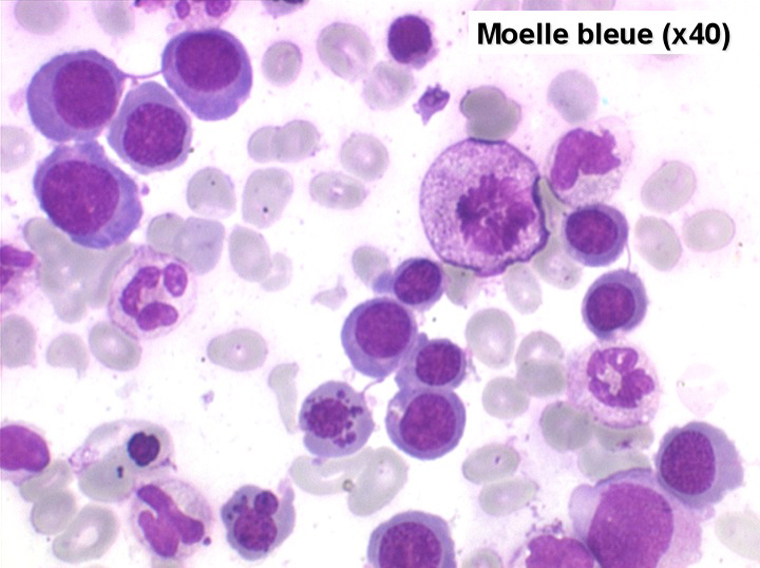
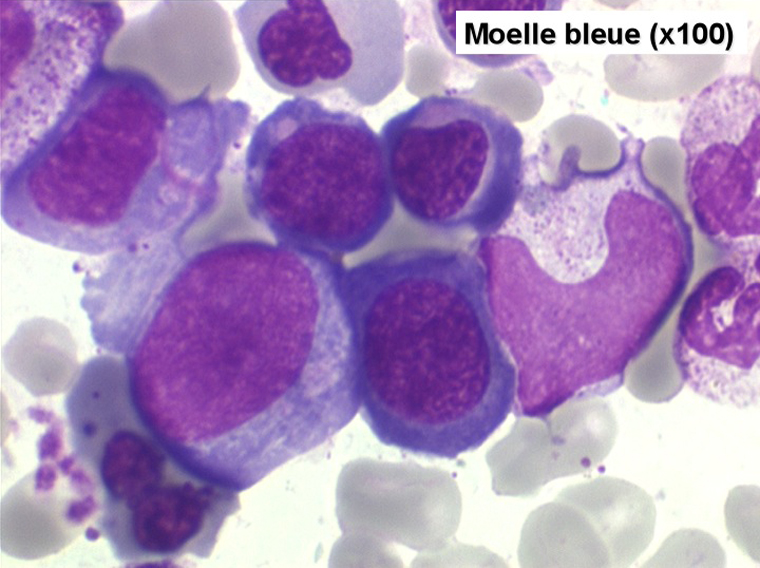
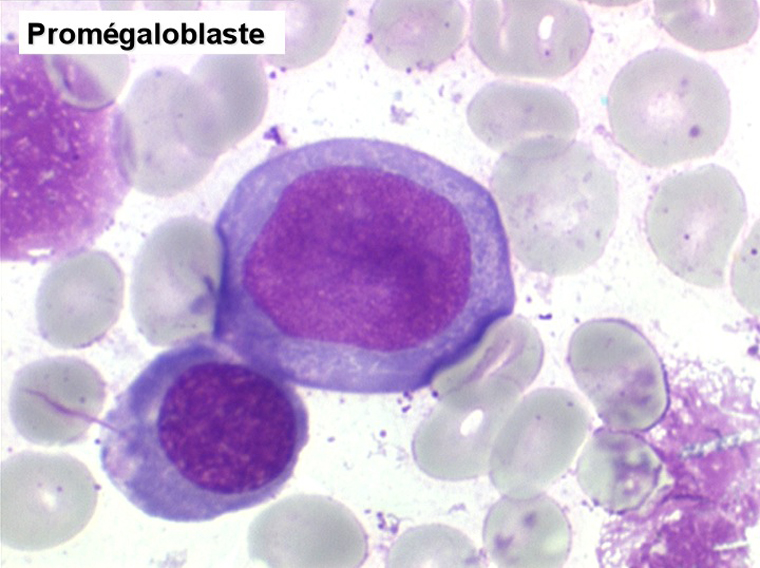
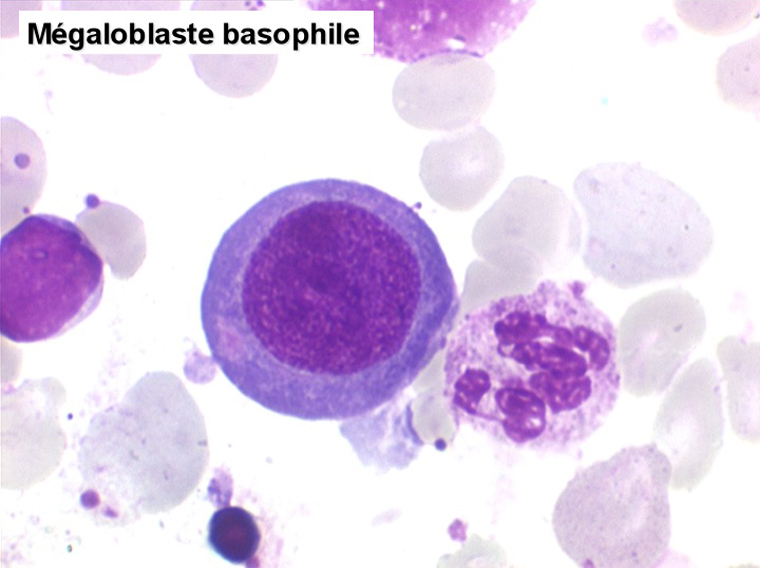
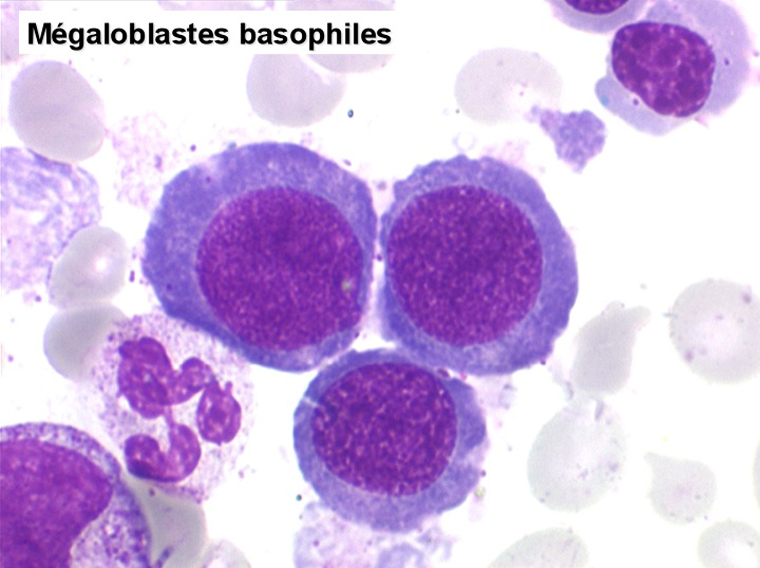
Le tableau hématologique est celui d'une myélodysplasie intense avec un gigantisme cellulaire (mégaloblastose), des polynucléaires hypersegmentés, des inclusions érythrocytaires multiples, une grande érythroblastose médullaire dystrophique (moelle « bleue »), et des anomalies morphologiques et fonctionnelles de l'ensemble des lignées médullaires. Cette myélodysplasie régresse, rapidement et totalement, par apport de vitamine B12 en injection intramusculaire.
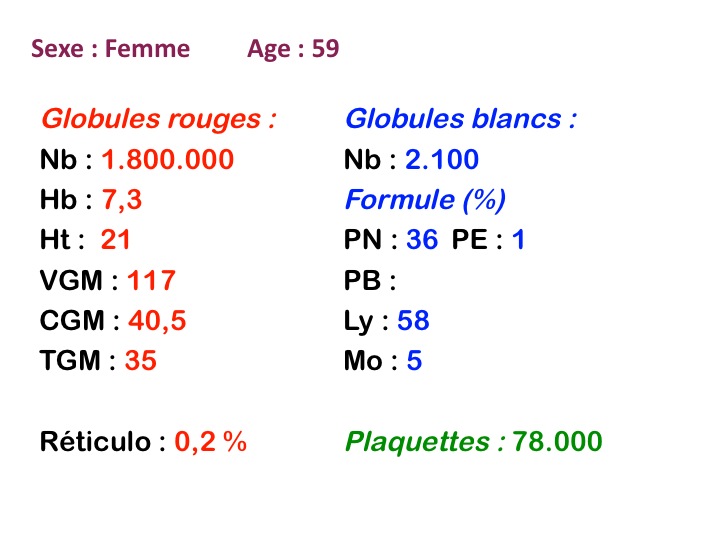
Sang de maladie de Biermer
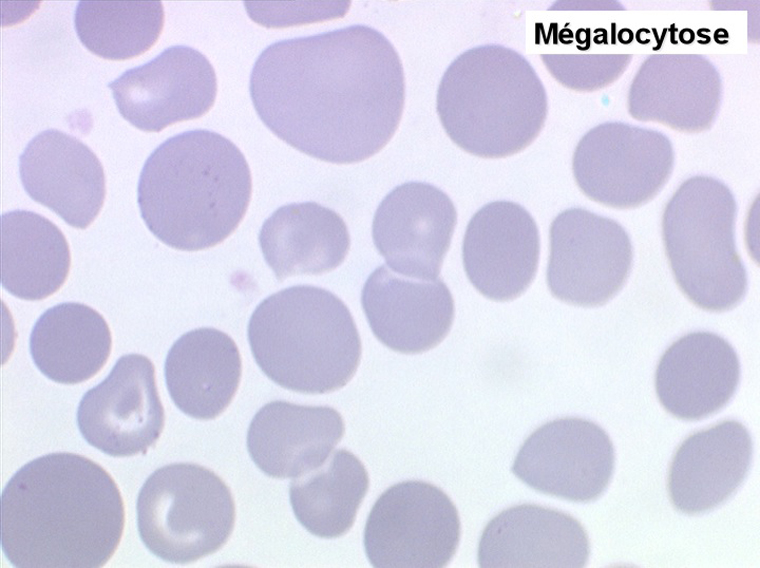
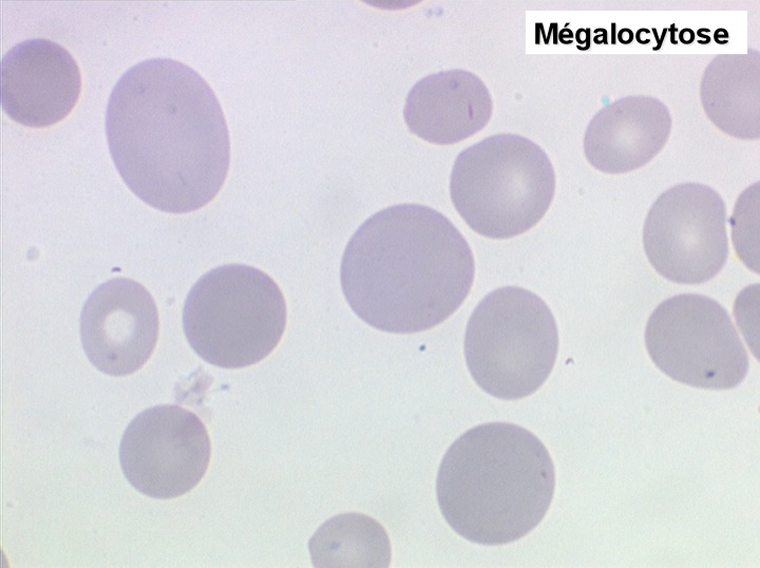
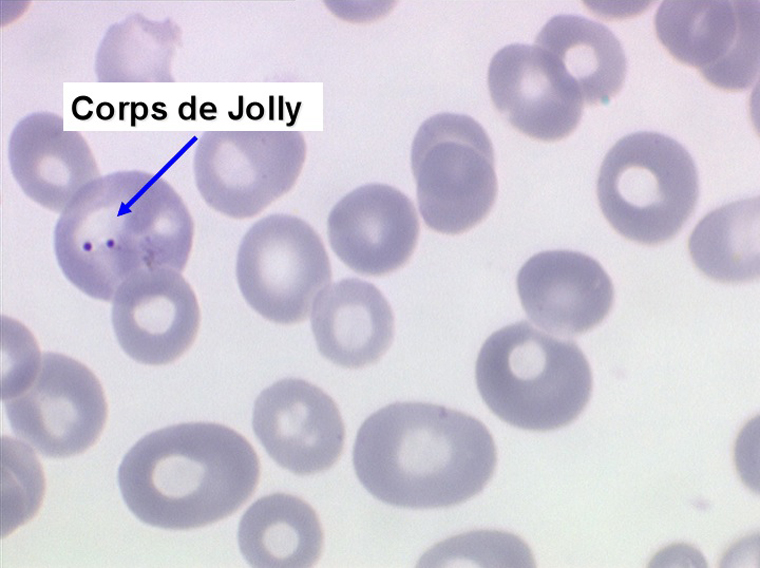
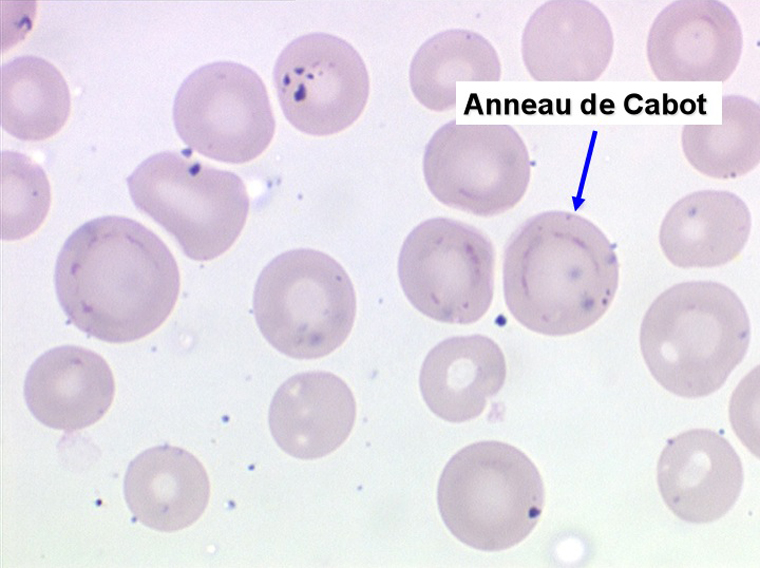
Moelle de maladie de Biermer
(4) – D'autres causes de myélodysplasie méritent une mention :
• Le saturnisme (intoxication par le plomb) est responsable d'une anémie hypochrome hypersidérémique mêlant blocage de synthèse de l'hémoglobine et hémolyse. La présence sur le frottis de sang d’hématies contenant des ponctuations basophiles est habituelle. Le taux de ces hématies ponctuées est proportionnel à l’intensité de l’intoxication.
• L'hypersplénisme, quelque soit la cause de la splénomégalie mais surtout s'il s'agit d'une splénomégalie inflammatoire, se traduit par un blocage médullaire et une pancytopénie à moelle riche. Dans ce cas l'anémie peut être relativement régénérative car la rate elle-même, organe macrophage par excellence, est hémolysante.
• Les anémies inflammatoires observées au cours de maladies infectieuses ou inflammatoires au long cours, sont de type microcytaire avec anomalies complexes du métabolisme du fer. Celles-ci se traduisent le plus souvent par une ferritine élevée alors que le fer sérique et la sidérophiline sont abaissés.