Lymphocytes
3 – Syndromes lymphoprolifératifs
Les syndromes lymphoprolifératifs (SLP) sont des hyperlymphocytoses sanguines malignes, caractérisées par une augmentation de cellules lymphoïdes dans le sang. On en distingue deux groupes. L'un est fait de cellules lymphoïdes « jeunes » et correspond aux leucémies aiguës lymphoblastiques. L'autre est fait de cellules lymphocytaires « mûres » et correspond à la leucémie lymphoïde chronique et à ses états frontières.
1 – Les leucémies aiguës lymphoblastiques (LAL)
Les leucémies aiguës lymphoblastiques s'observent surtout, mais pas exclusivement, chez l'enfant. Leur diagnostic est évoqué sur l'examen de sang : une insuffisance médullaire associée à une blastose sanguine.

Les blastes ne sont pas granuleux et ne contiennent jamais de corps d'Auer. Cependant la seule cytologie morphologique ne suffit pas à affirmer la nature lymphoïde des blastes et, actuellement, la classification des LAL est fondée uniquement sur l'étude immunologique des blastes (antigènes de surface et cytoplasmiques). Cependant ce site étant dédié à la seule morphologie cellulaire nous décrirons les LAL selon l'ancienne classification FAB qui a le mérite de la simplicité. La classification FAB des leucémies aiguës lymphoblastiques comporte 3 types de LAL, selon la morphologie des blastes.
(1) – LAL 1
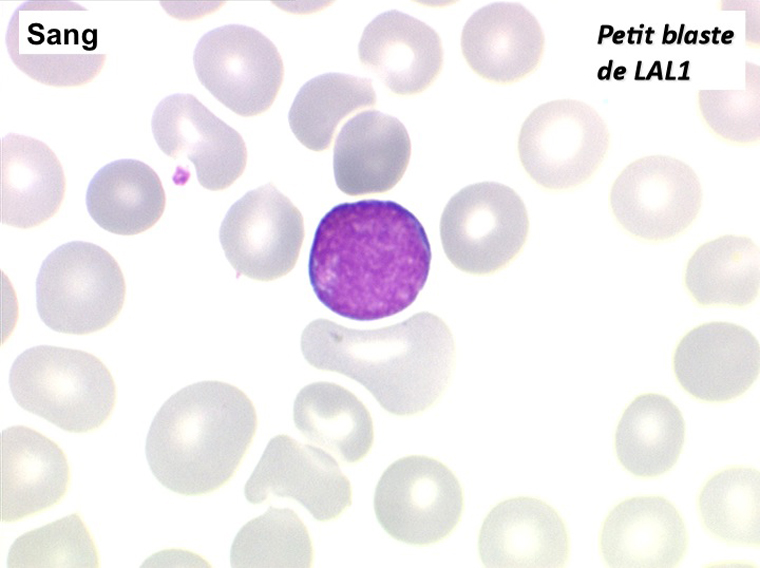
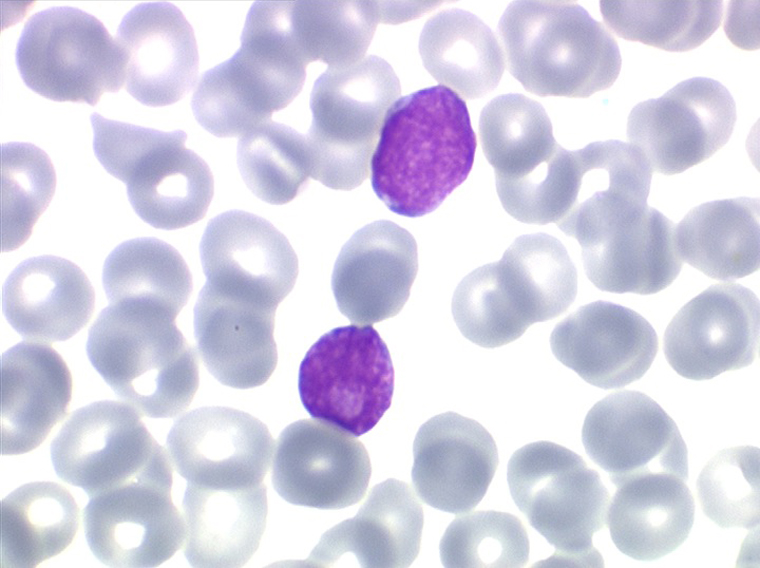
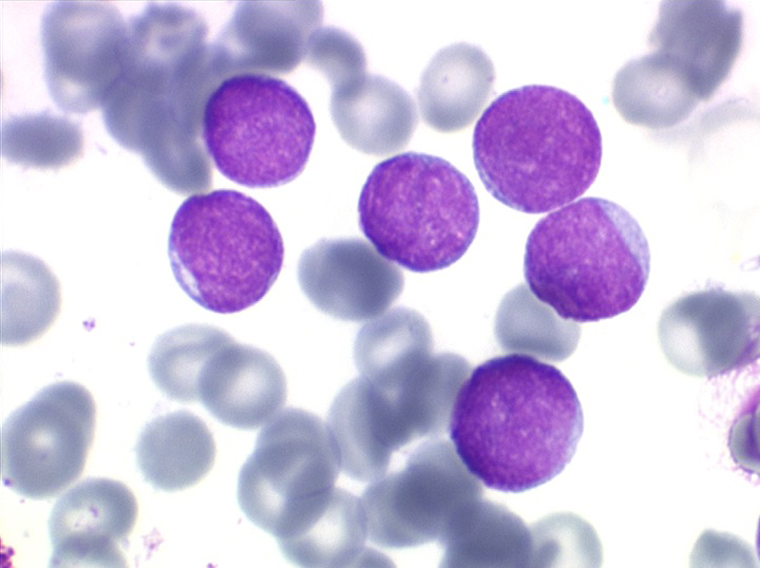
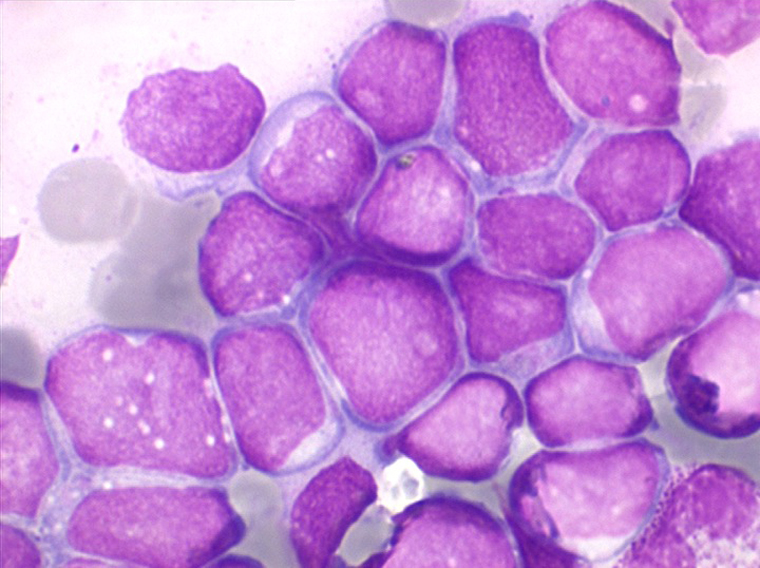
Les blastes, non granuleux, sont de petite taille, à rapport nucléo-plasmatique élevé, avec un seul nucléole pâle souvent non visible. Il peut y avoir un mélange de petits blastes et de blastes plus grands, mais le pourcentage de petits blastes doit rester supérieur à 75%. C'est la forme habituellement observée chez l'enfant. Lorsque les blastes sont tous de petite taille, à nucléole pas ou peu visible, le diagnostic avec des lymphocytes mûrs n'est pas toujours facile chez l'enfant, mais celui-ci ne faisant jamais de leucémie lymphoïde chronique toute hyperlymphocytose monomorphe doit chez lui être considérée comme suspecte. À une exception près cependant, la coqueluche, qui peut, précocement et avant la période des quintes, être responsable d'une hyperlymphocytose mûre et monomorphe atteignant 20.000 à 30.000/µl ou plus, mais, critère essentiel, cette hyperlymphocytose monomorphe ne s'accompagne pas d'insuffisance médullaire (ni anémie, ni thrombopénie).
Sang de LAL1
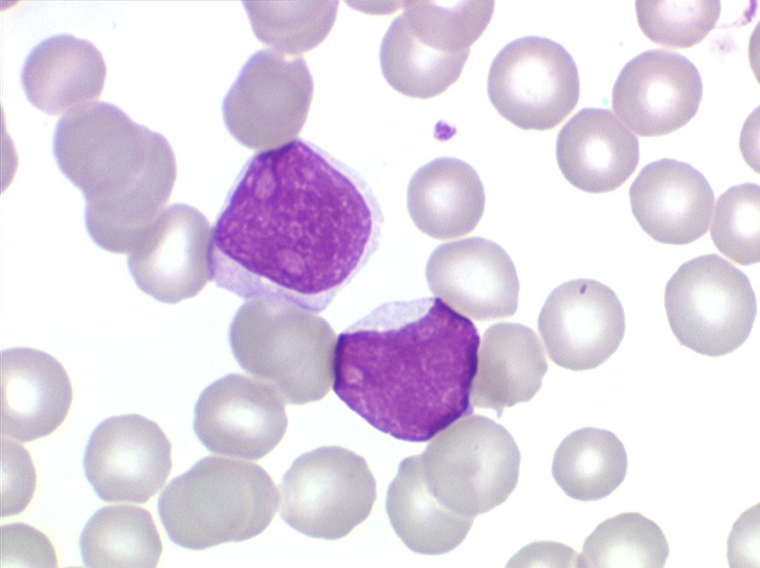
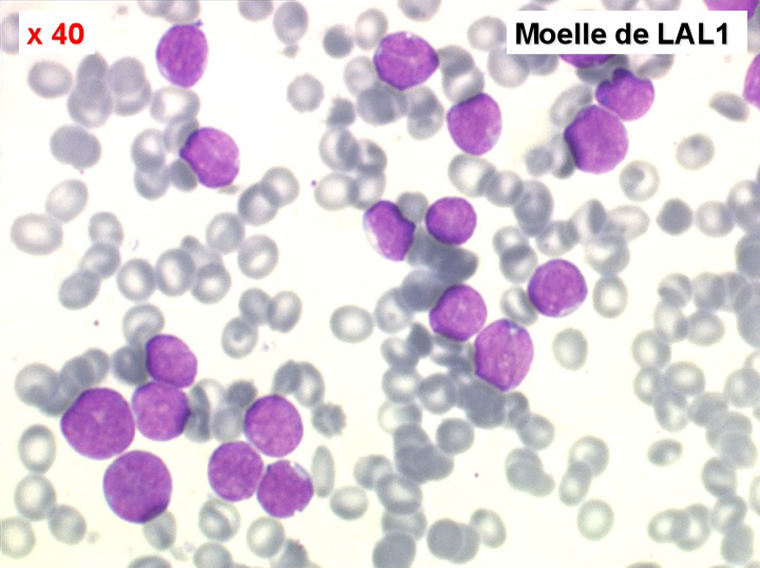
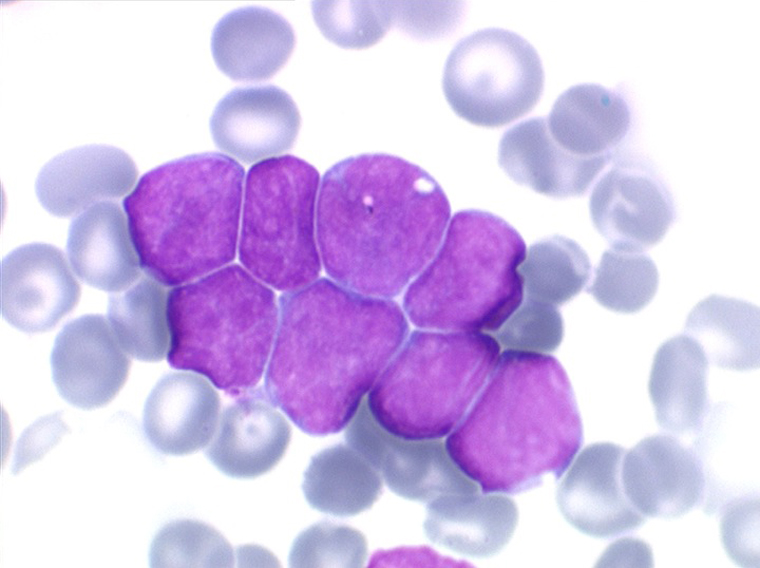
Moelle de LAL1
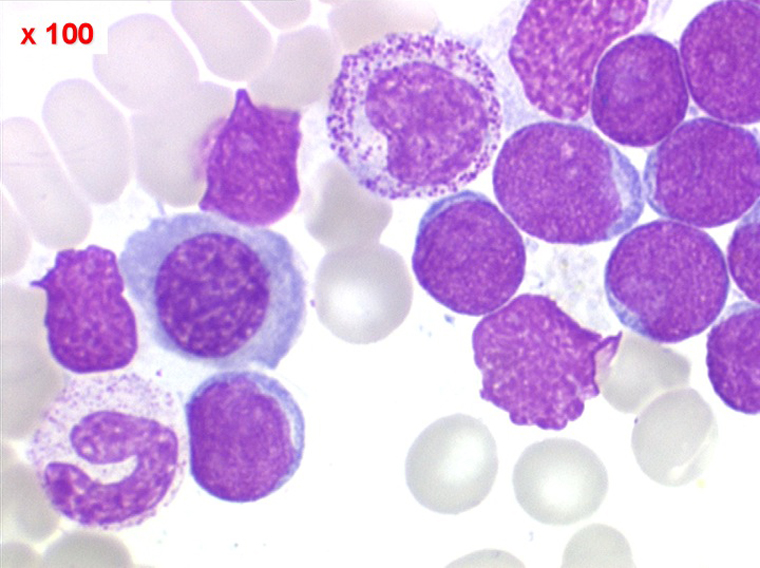
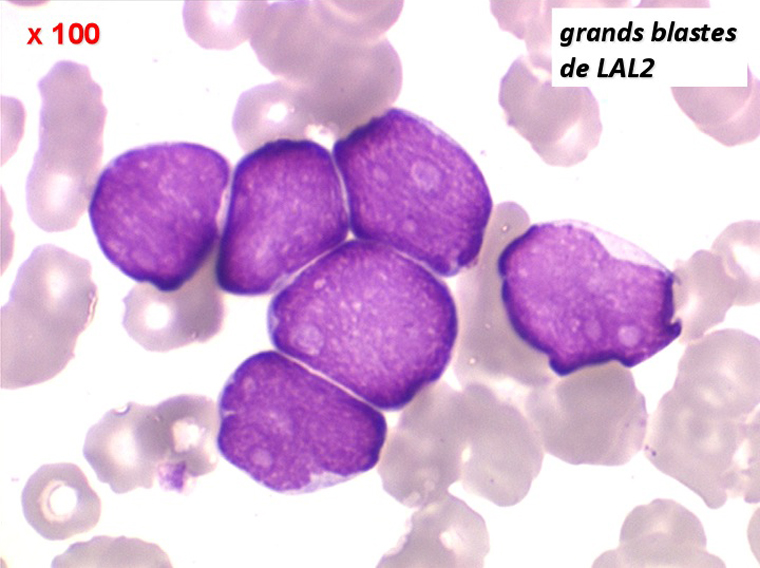
(2) – LAL 2
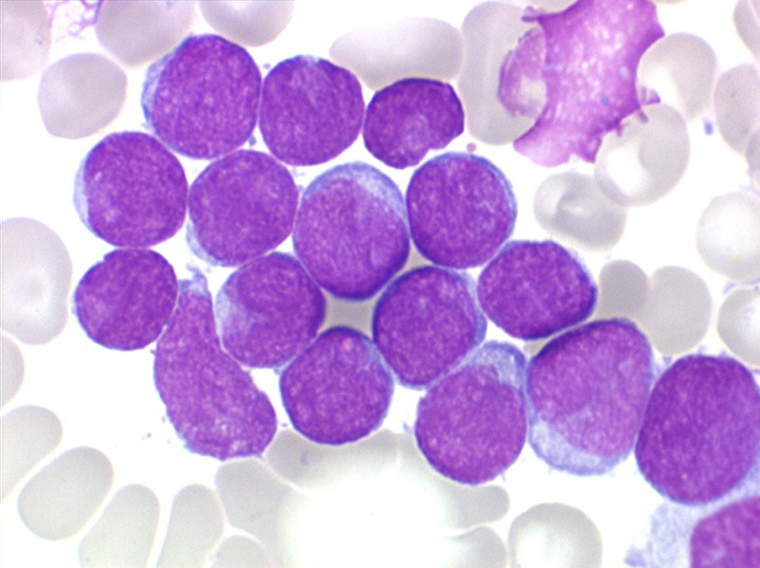
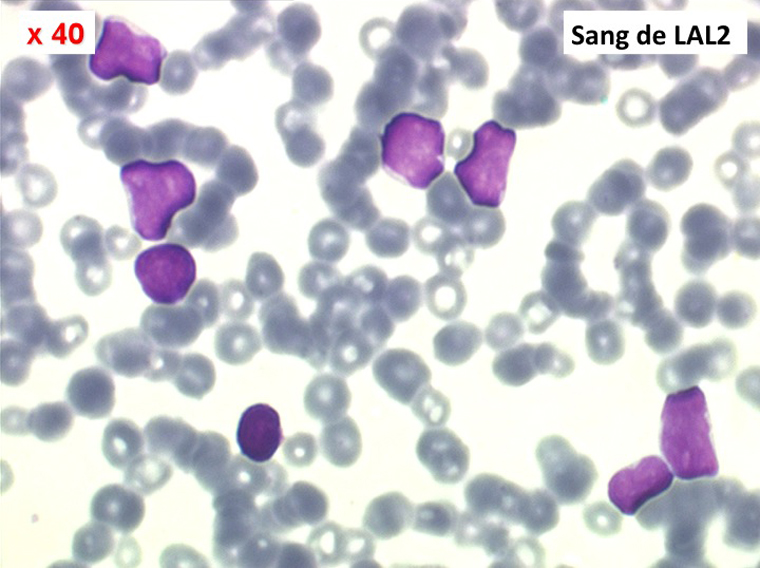
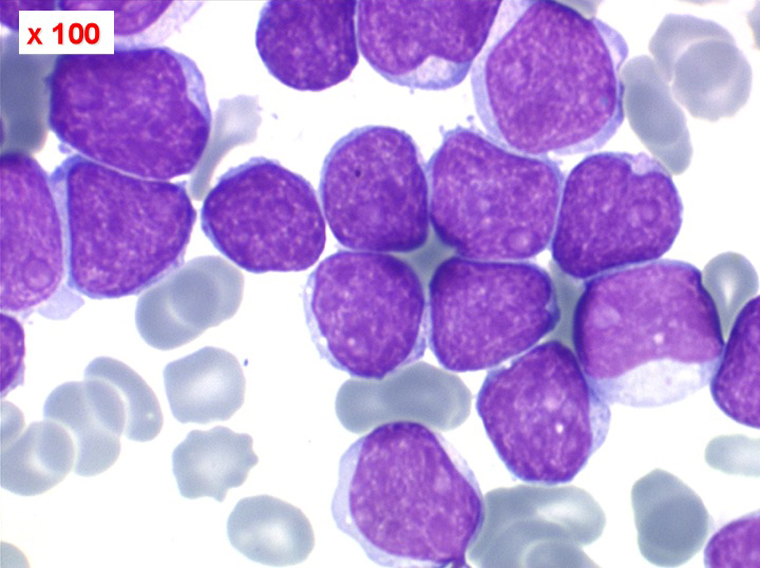
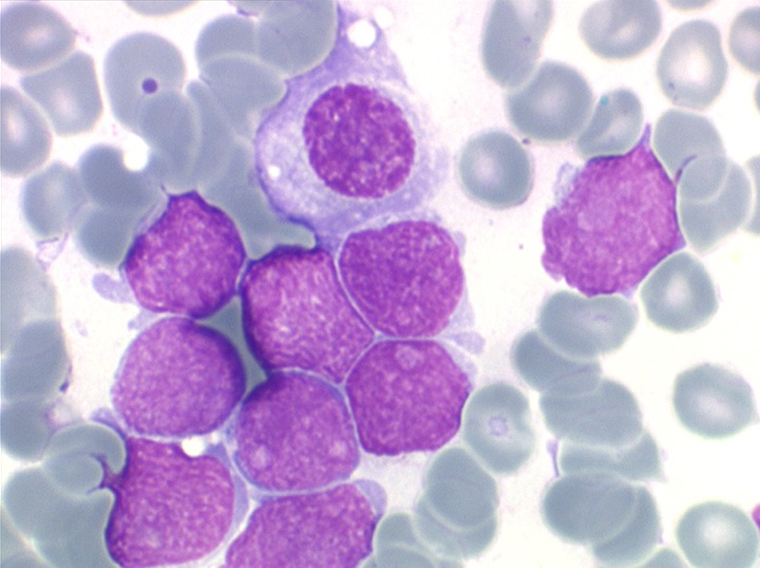
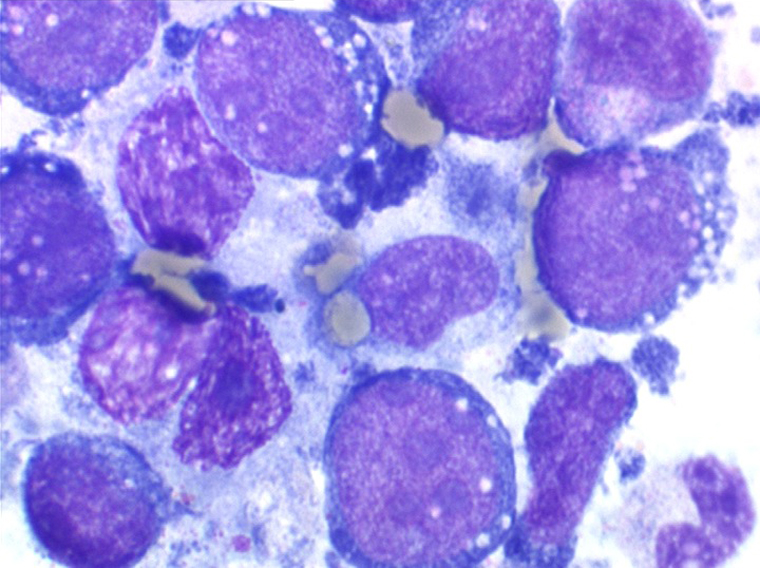
Les blastes, non granuleux, sont de grande taille, plus du double du lymphocyte normal, le rapport nucléo-plasmatique est variable mais élevé dans un grand nombre de blastes, les nucléoles sont multiples, en nombre variable, habituellement bien visibles. C'est la forme habituelle des LAL de l'adulte.
Sang de LAL2
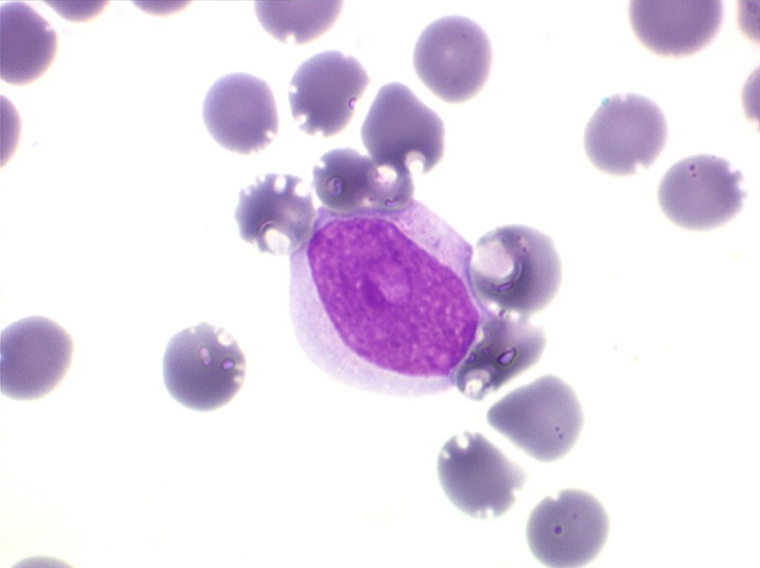
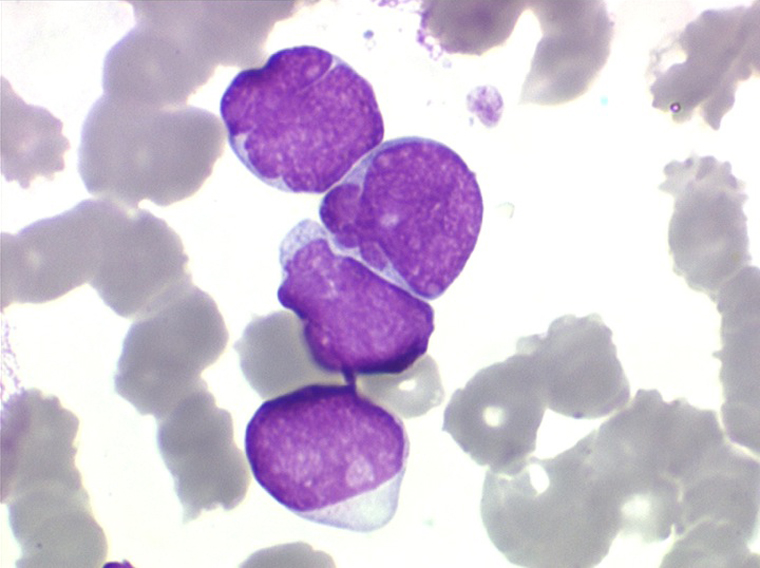
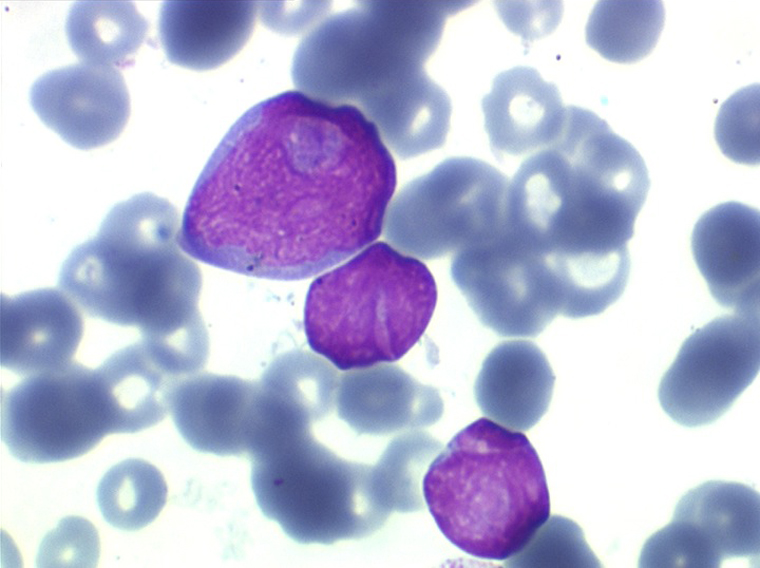
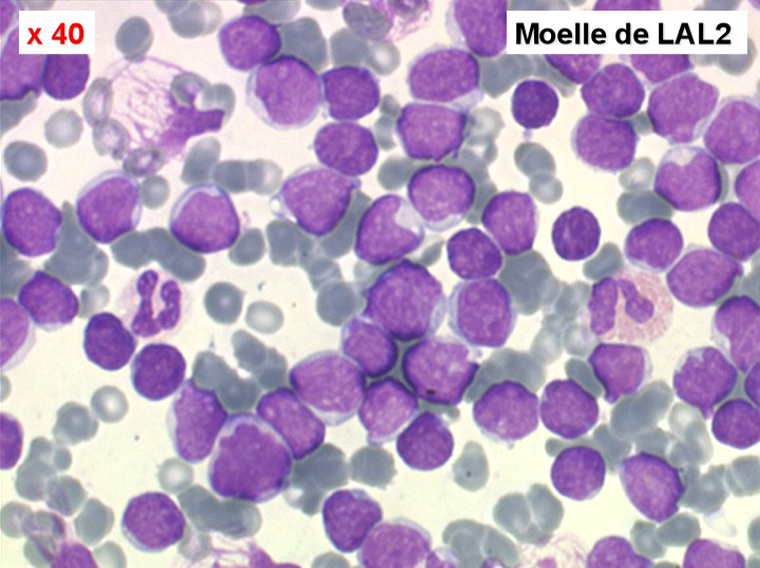
Moelle de LAL2
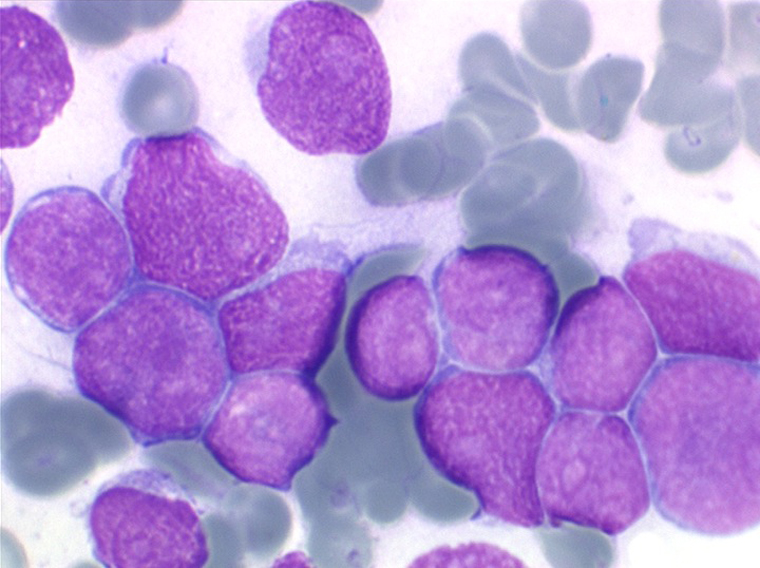
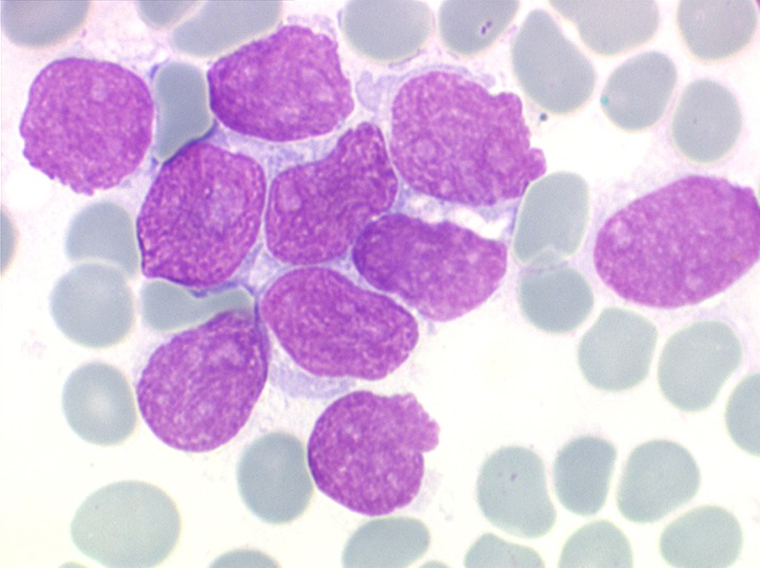
(3) – LAL 3
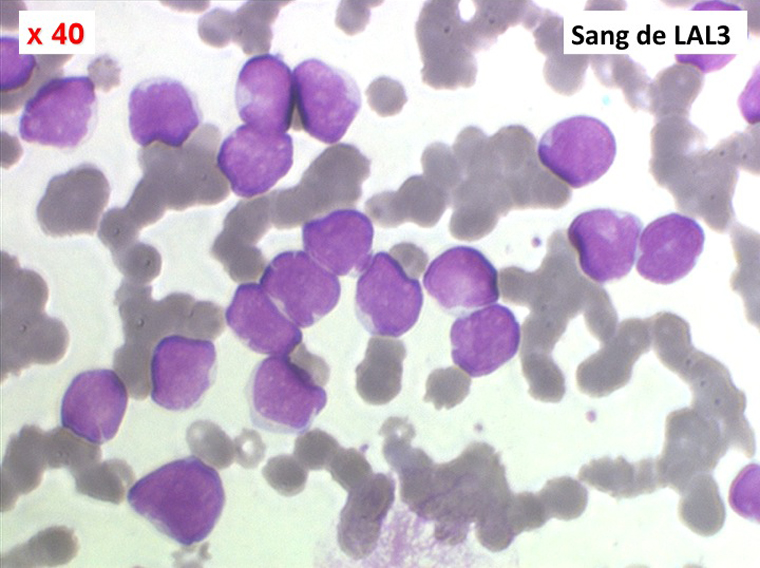
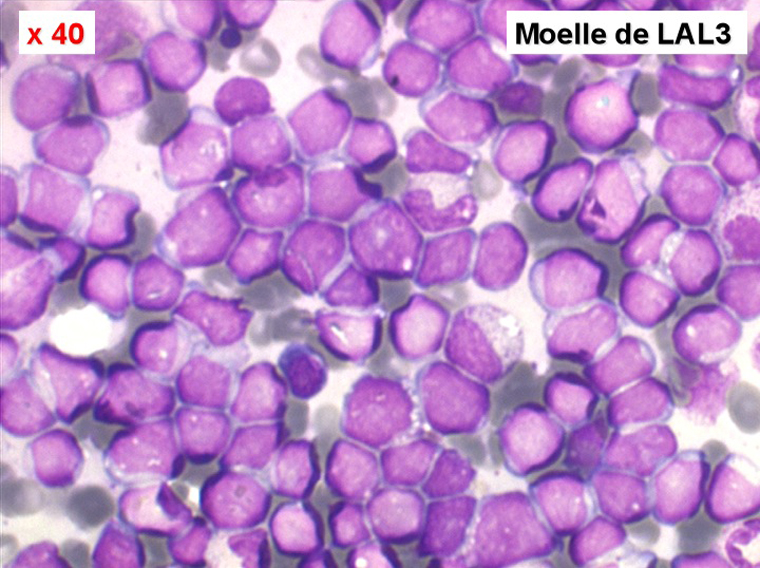
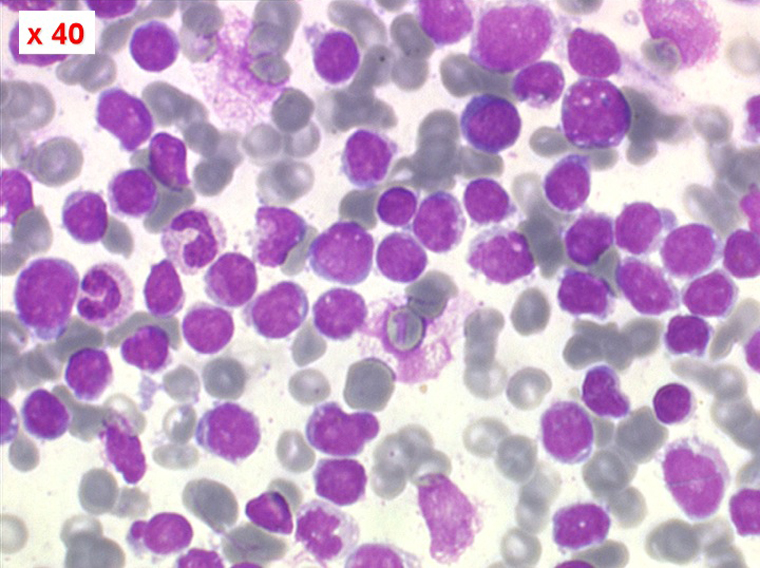
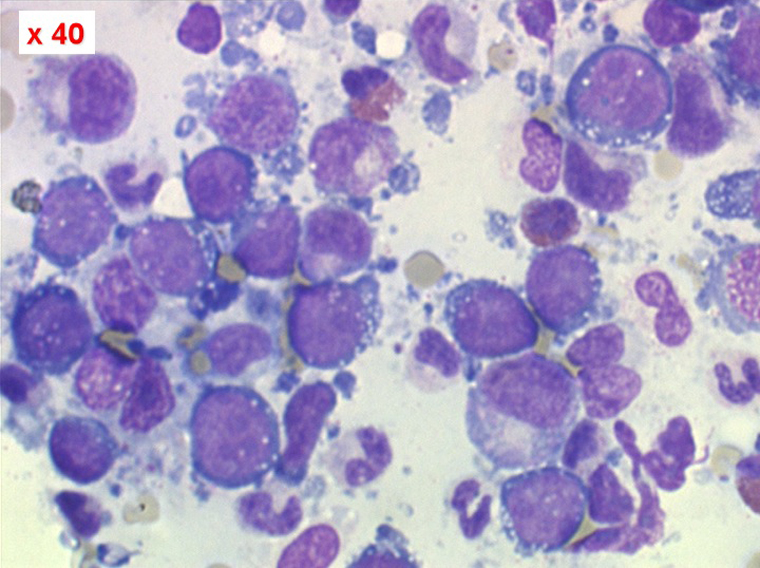
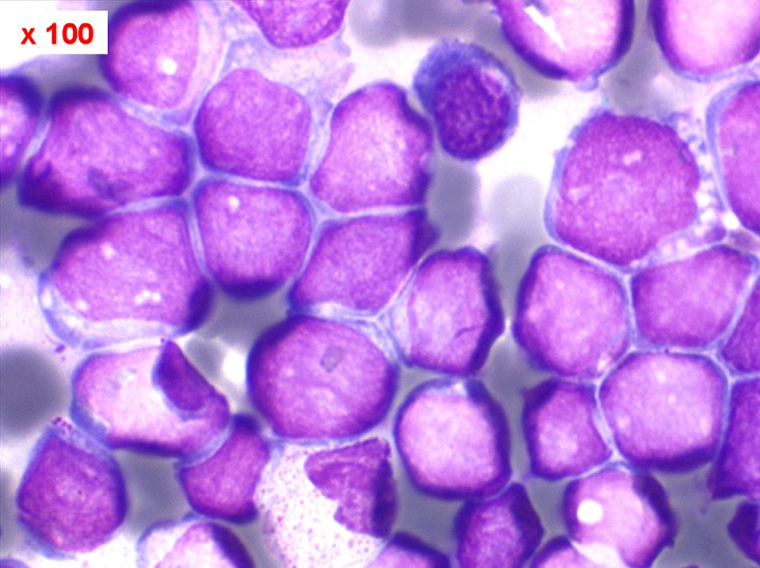
Les blastes ont la morphologie caractéristique des cellules tumorales du lymphome de Burkitt. Ils sont de taille moyenne ou grande, véritables immunoblastes à cytoplasme abondant hyperbasophile, à rapport nucléo-plasmatique élevé, avec un noyau contenant des nucléoles très visibles, et surtout la présence dans un grand nombre de blastes de petites vacuoles dans le noyau et dans le cytoplasme. Ces blastes sont souvent fragiles, éclatés, entourés de dépôts d'immunoglobulines, surtout dans la moelle.
C'est une leucémie particulièrement grave, touchant des adolescents et des adultes jeunes, envahissant très vite de nombreux organes, franchissant les barrières méningées et gonadiques.
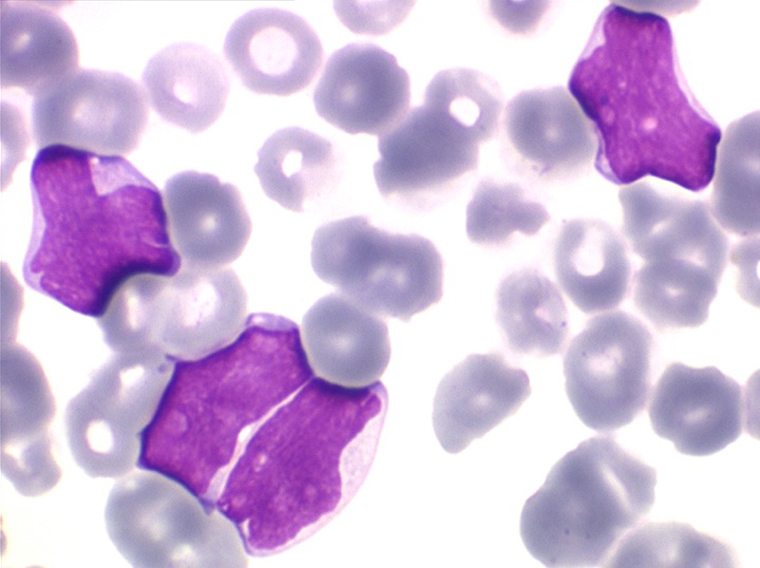
Sang de LAL3
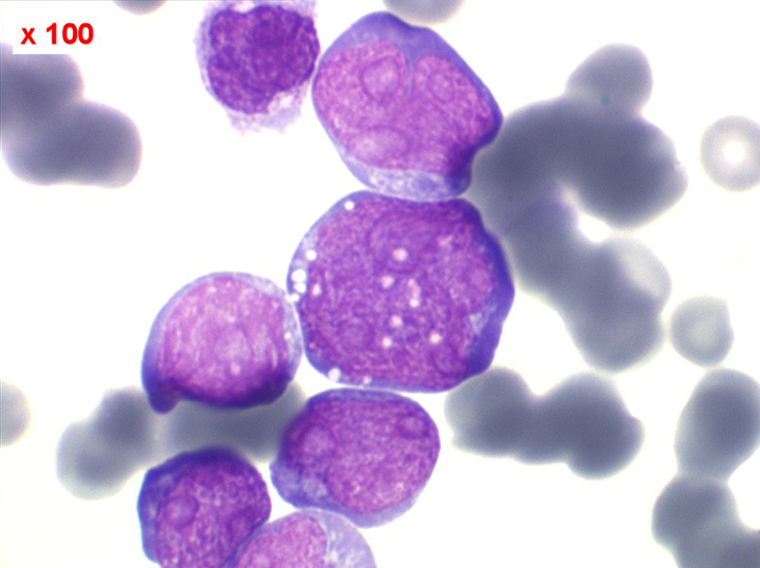
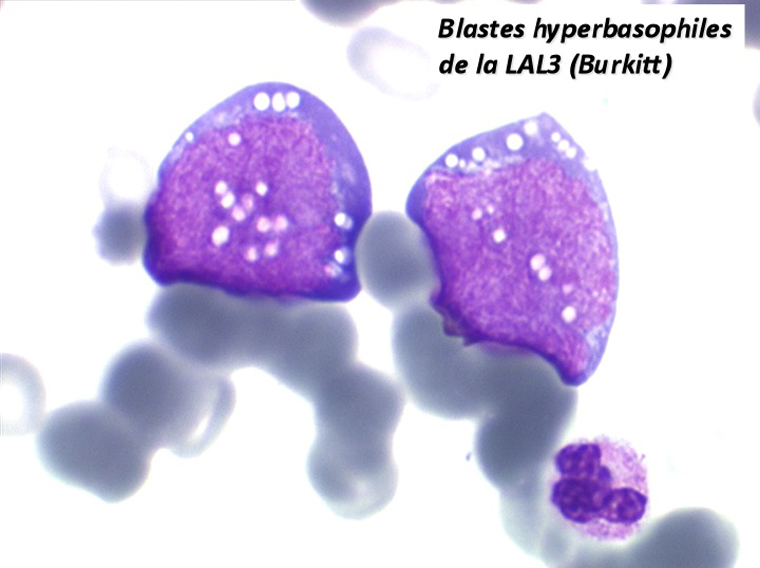
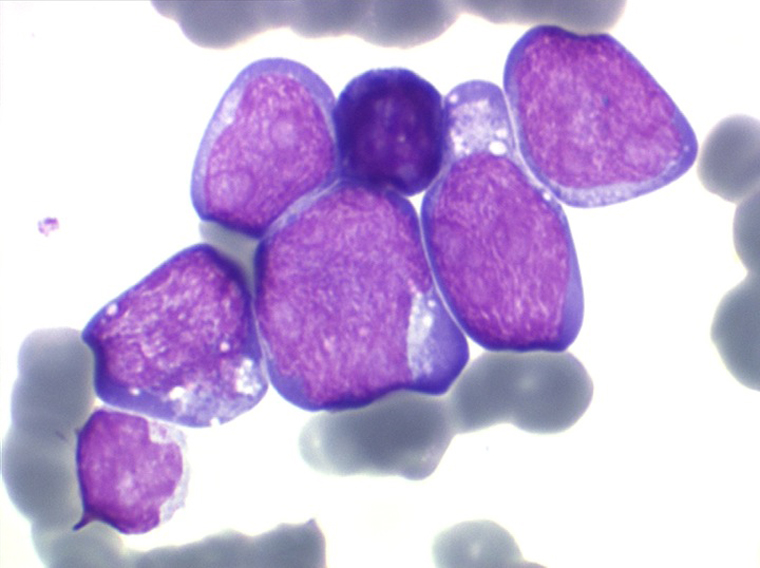
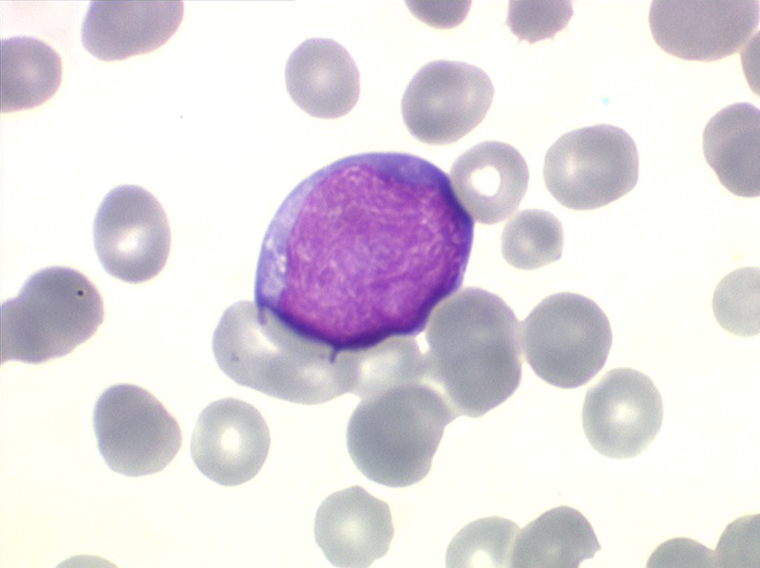
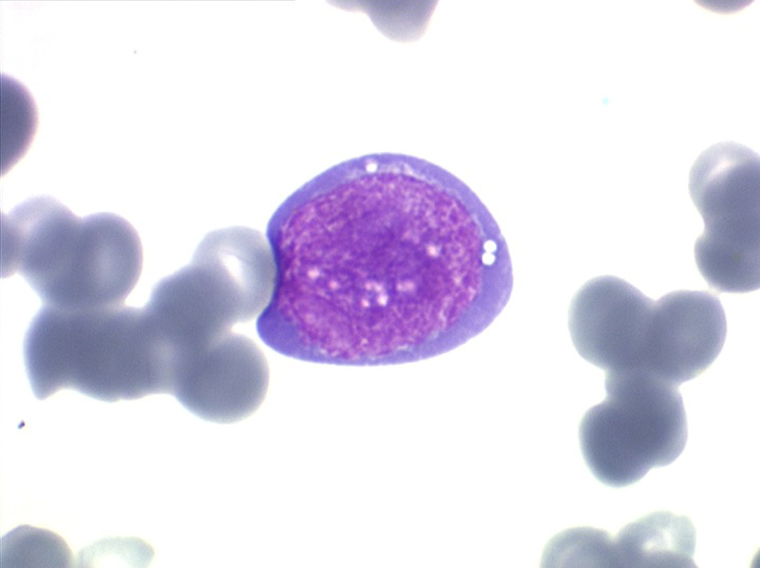
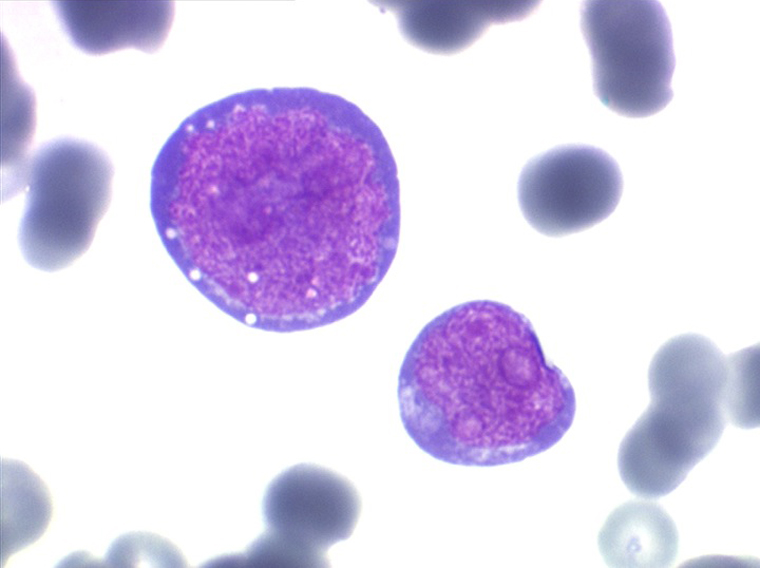
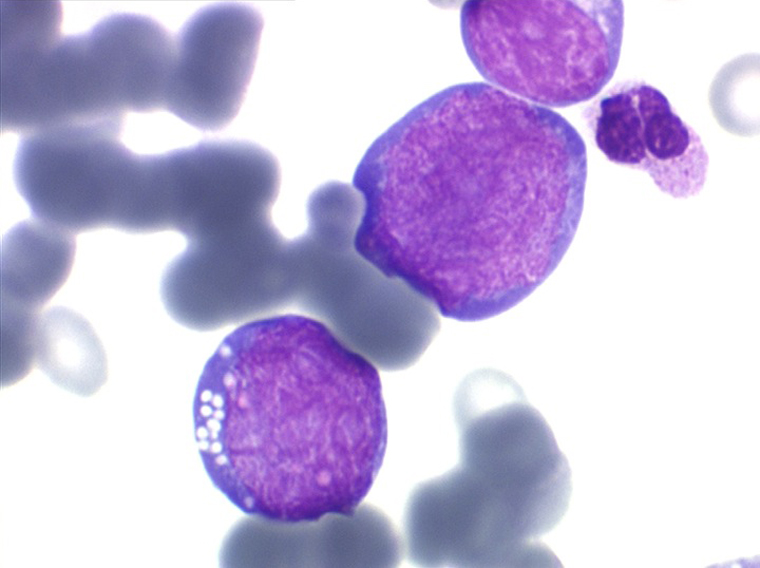
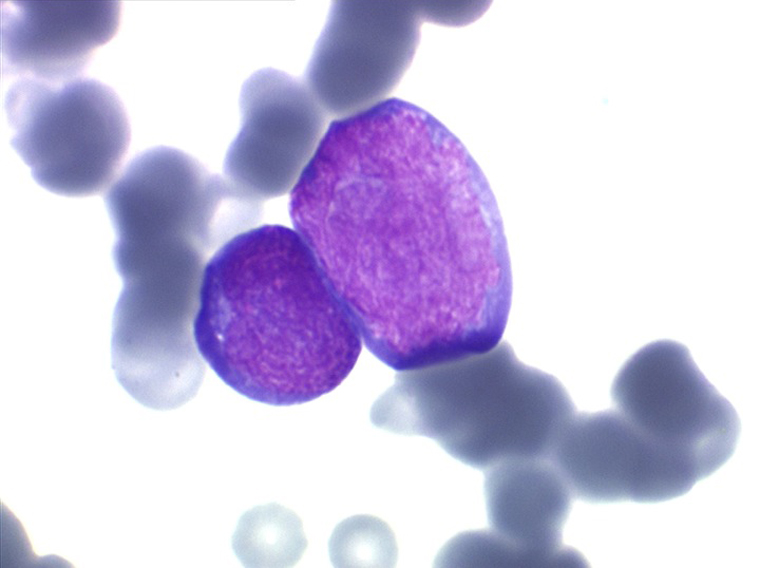
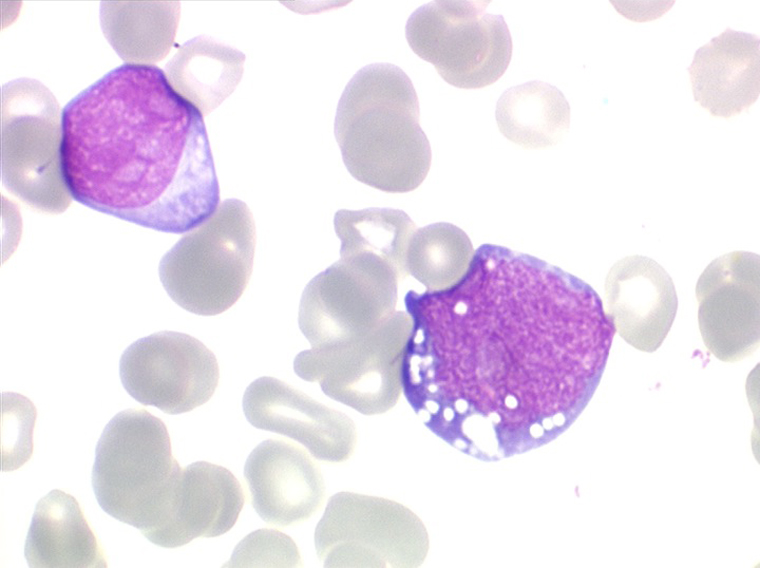
Moelle de LAL3
Il convient de compléter cette description morphologique des LAL par deux remarques :
• Les LAL1 et LAL2 sont souvent hyperleucocytaires, et le degré de cette hyperleucocytose est corrélé au pronostic.
• Il peut y avoir dans le sang une discrète myélémie ou érythromyélémie qui ne doit pas induire en erreur et faire conclure à tort que les blastes non granuleux sont de nature myéloïde. La myélémie, commune à tous les processus de métastase médullaire, est due au fait que l'infiltration lymphoblastique de la moelle osseuse se comporte comme un envahissement de cellules étrangères.
2 – La leucémie lymphoïde chronique et les états frontières
Outre la leucémie lymphoïde chronique, la plus fréquente, ce groupe de syndromes lymphoprolifératifs comprend la leucémie à prolymphocytes et la leucémie à tricholeucocytes.
(1) – La leucémie lymphoïde chronique (LLC)
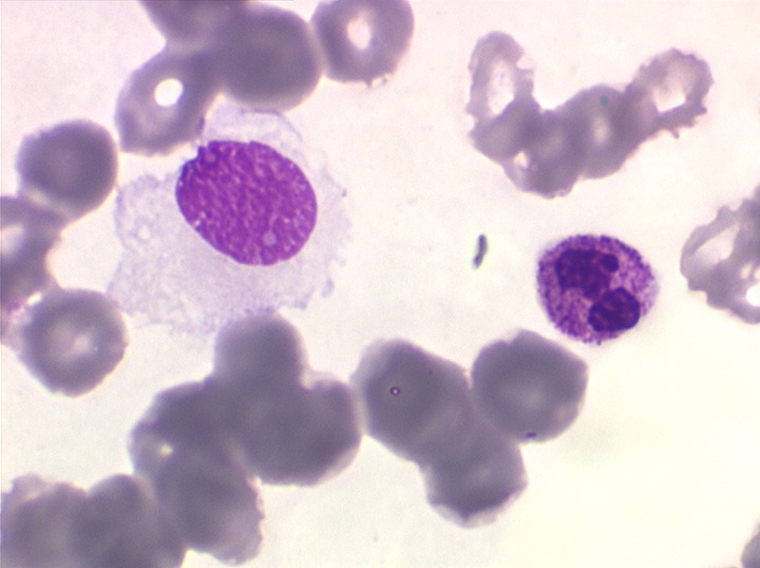
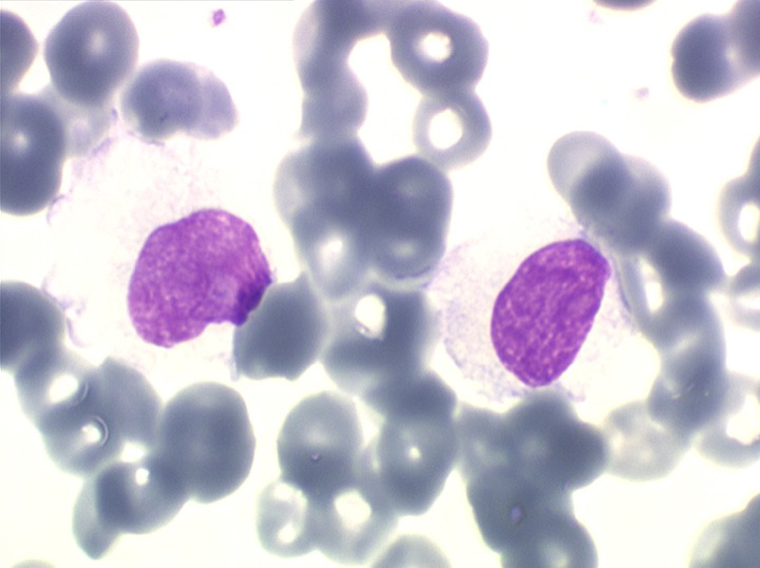
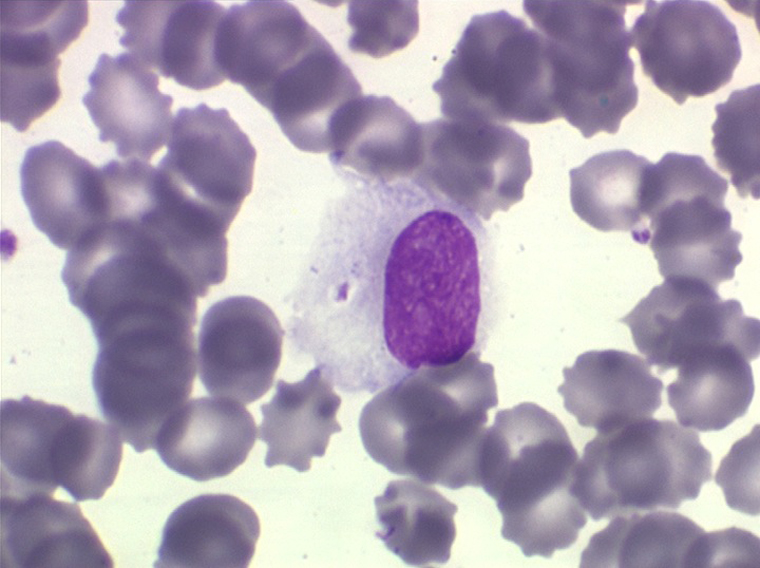
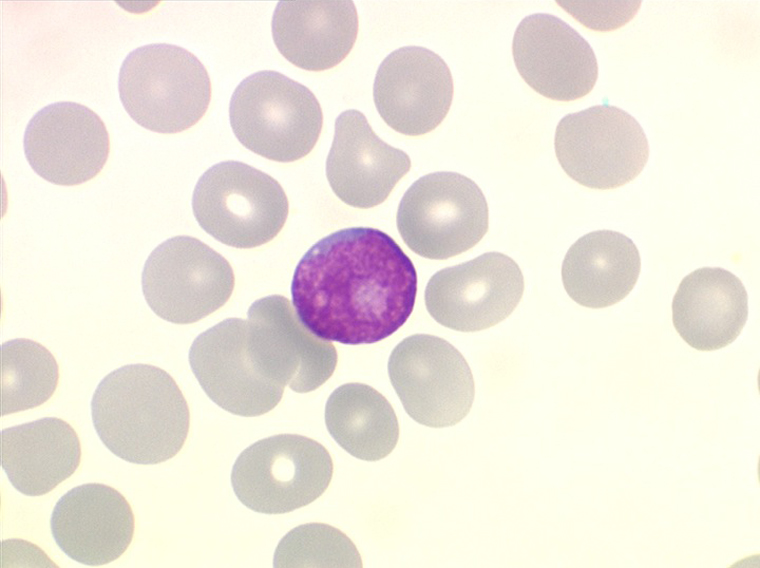
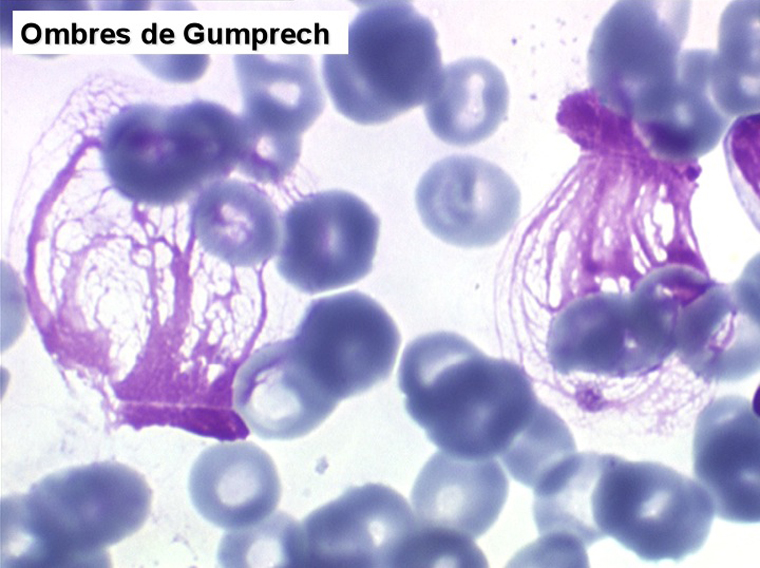
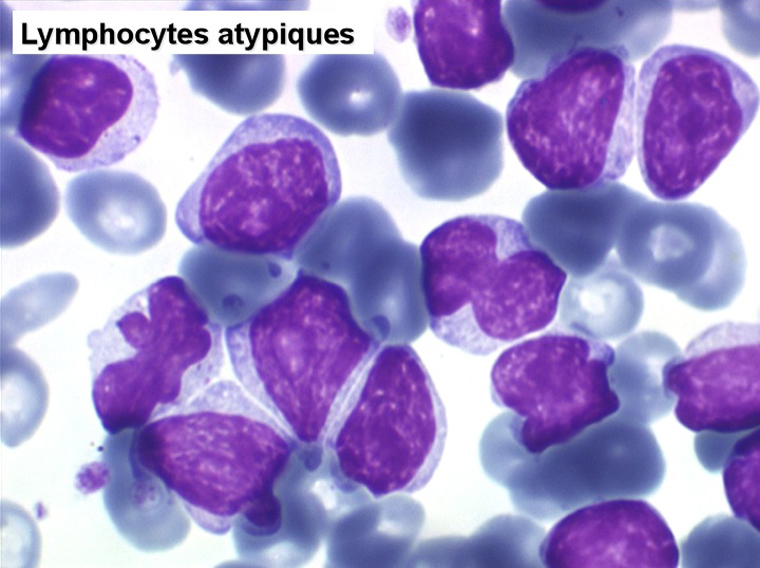
La LLC est caractérisée par une hyperlymphocytose sanguine monomorphe, supérieure à 5.000/µl, habituellement beaucoup plus, faite de lymphocytes mûrs, de taille petite ou moyenne. Il y a une infiltration lymphoïde diffuse de la moelle osseuse représentant plus de 30% des cellules, mais celle-ci est bien tolérée et la NFS ne montre pas de stigmate d'insuffisance médullaire, sauf en phase terminale. Dans la moelle les lymphocytes ont souvent un aspect plus jeune que dans le sang, avec une chromatine laquée et des nucléoles. Sur la lame de sang certains lymphocytes sont abimés, leur noyau dilacéré seul visible sous forme d'une résille (les ombres de Gumprech) à qui on a voulu, à tort, faire jouer un rôle pronostique. Ces ombres de Gumprech ne sont pas spécifiques de LLC et s'observent au cours d'autres syndromes lymphoprolifératifs, notamment les LAL.
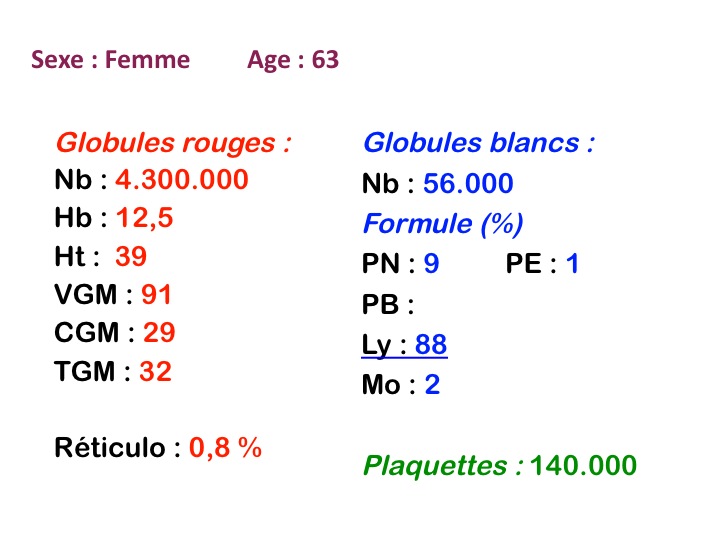
Cette hyperlymphocytose est monoclonale comme le prouve l'immunophénotypage des cellules leucémiques qui montre qu'elles sont porteuses de la même immunoglobuline de surface avec la même chaîne légère. Cet examen, devenu courant, évite de faire un myélogramme. Le fait important pour l'évolution de la maladie est que cette hyperlymphocytose monoclonale (les lymphocytes proliférants ne savent reconnaître qu'un seul antigène) induit un véritable déficit de l'immunité humorale. Le taux des IgM est d'emblée abaissé et celui des IgG va progressivement et inexorablement baisser, au dessous de 3 g/l le risque infectieux devient inéluctable et est la cause du décès dans 75% des cas.
Cette maladie atteint des adultes de plus de 50 ans, ne se voit jamais chez l'enfant, s'accompagne ou non d'organomégalies (adénopathies, hépato-splénomégalie) et a une évolution chronique de plusieurs années. Le déficit immunitaire explique certes le risque infectieux mais explique aussi un risque de maladies auto-immunes (notamment une anémie hémolytique), de cancers, de lymphomes secondaires, de dysglobulinémie monoclonale associée. En revanche la LLC ne se termine jamais en leucémie aiguë.
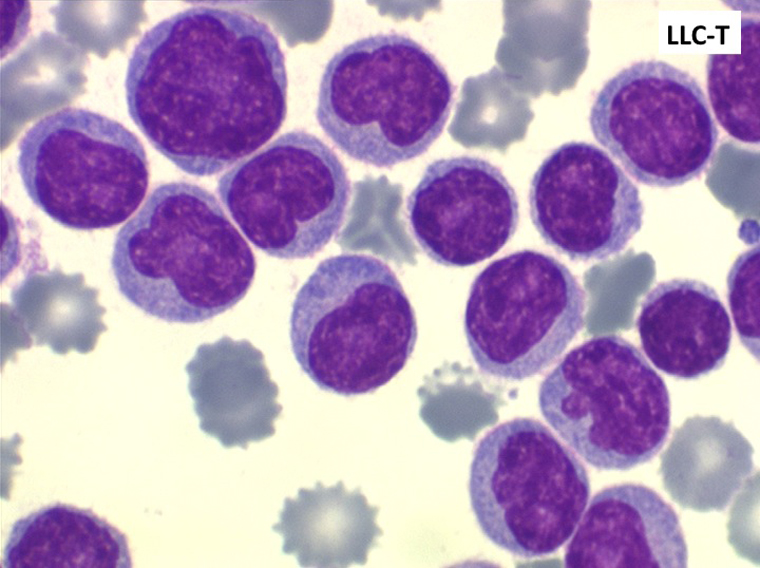
La plupart des LLC (>90%) sont de type B-lymphocytaire. Un petit nombre est fait de lymphocytes ayant des marqueurs T, leur pronostic est moins bon. Dans ce dernier cas les lymphocytes ont souvent un noyau irrégulier, dit convoluté. Si l'enfant ne fait jamais de leucémie lymphoïde chronique il faut rappeler qu'une hyperlymphocytose monomorphe peut être observée chez lui au cours de la coqueluche et poser un problème de diagnostic, non avec la LLC, mais avec une LAL1. Nous l'avons évoqué plus haut.
Sang de LLC
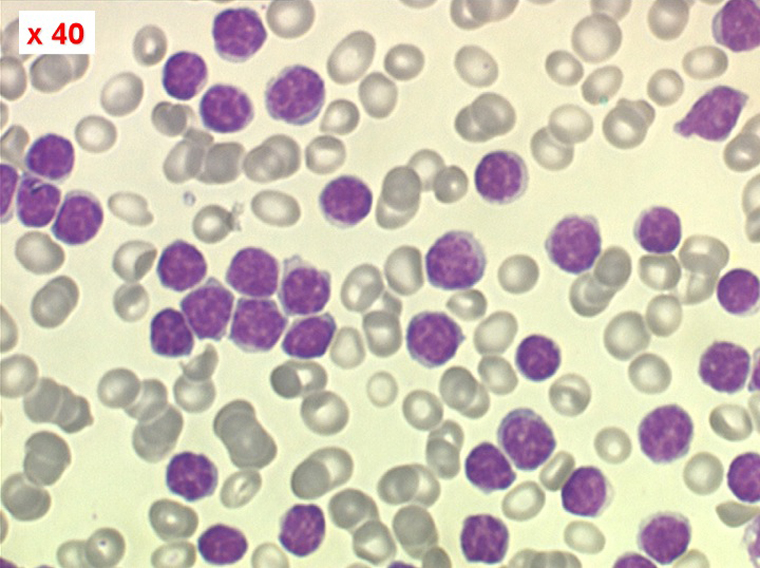
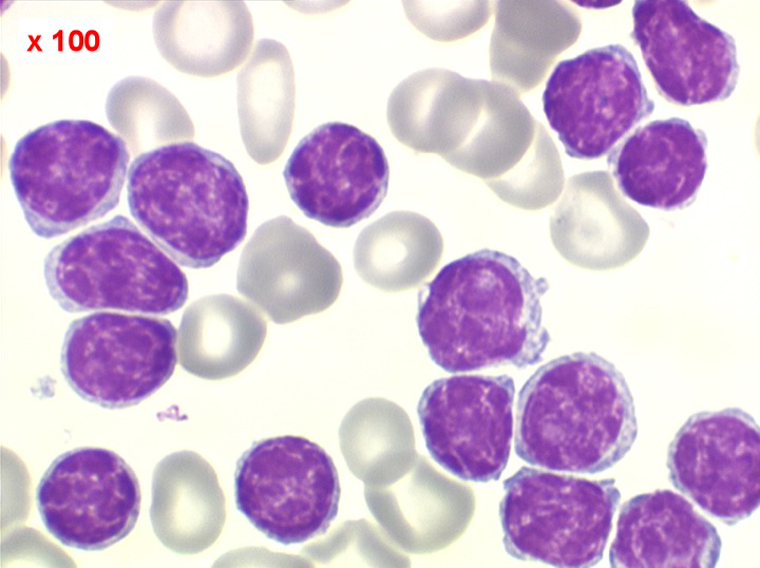
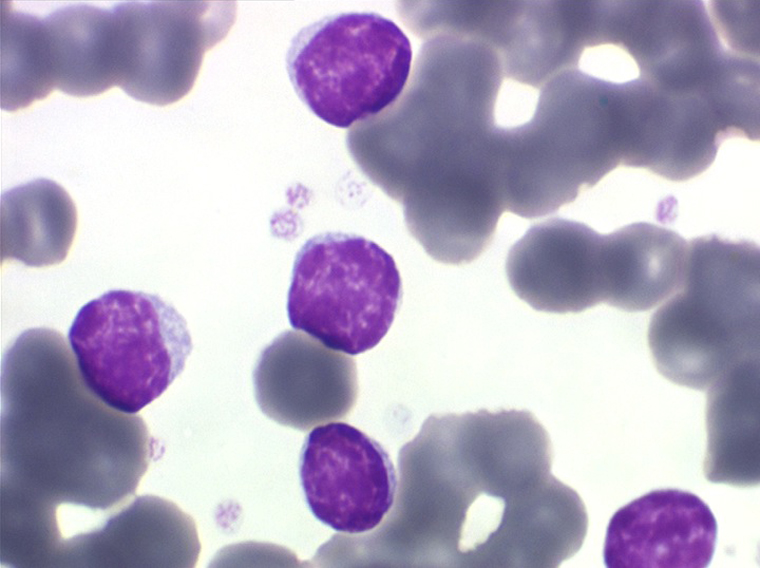
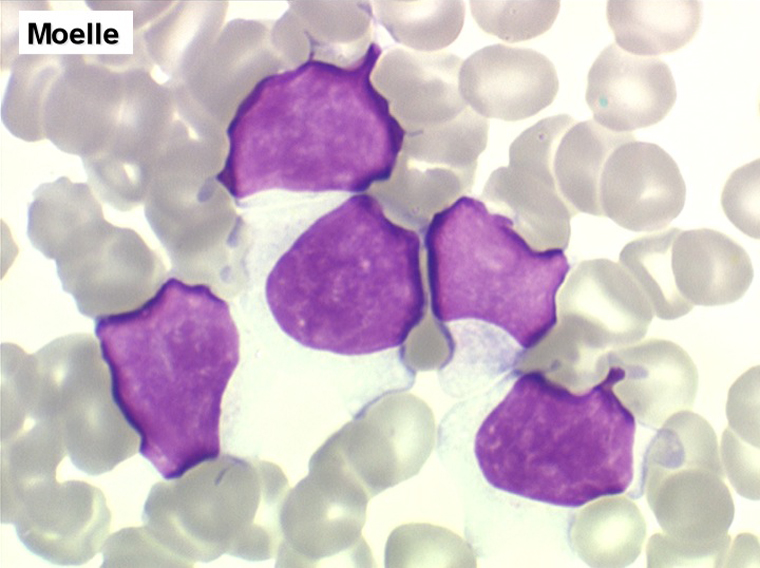
Moelle de LLC
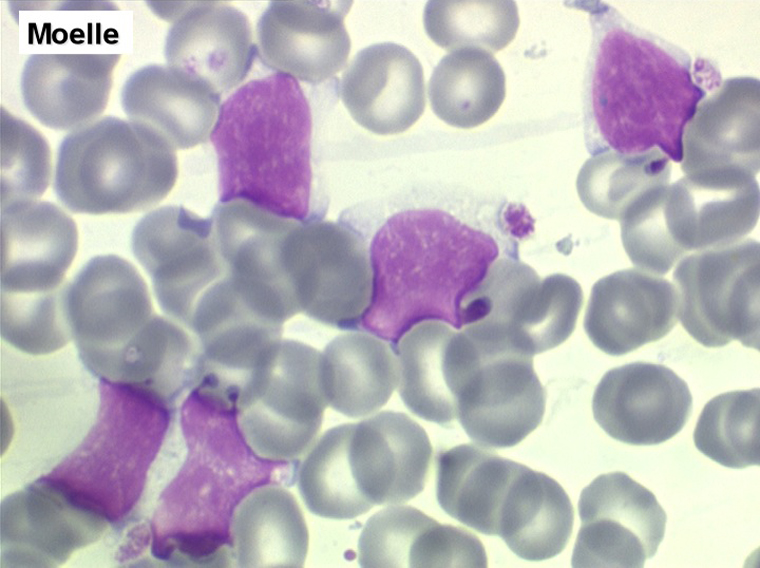
(2) – La leucémie à prolymphocytes (LPL)
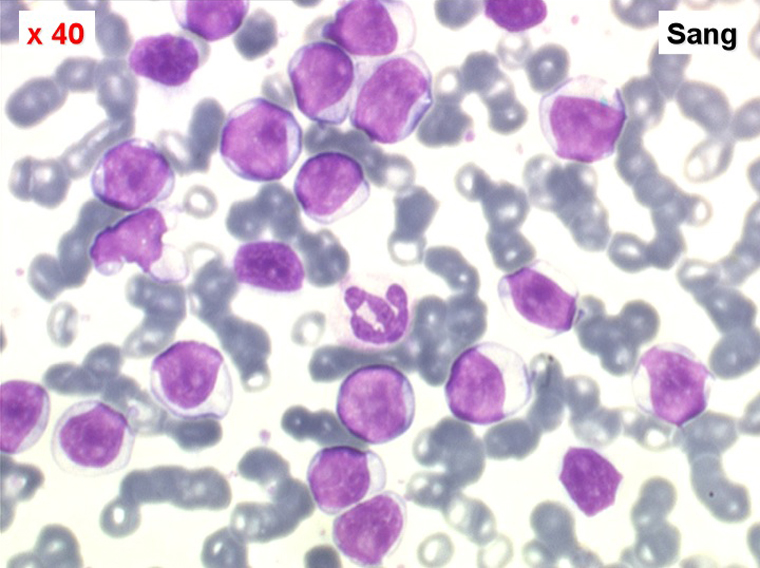
La LPL est aussi caractérisée par une hyperlymphocytose sanguine monomorphe, mais très importante (généralement >100.000/µl) et faite de prolymphocytes, grandes cellules rondes à cytoplasme bleu abondant, à noyau central possédant un gros nucléole lui aussi central donnant à la cellule un aspect de cible de tir. L'immunomarquage montre qu'il s'agit d'un lymphocyte « hyper-B » avec un marquage très important d'immunoglobulines de surface monoclonales. Cette maladie atteints des sujets âgés, donne une insuffisance immunitaire rapide, est chimiorésistante et d'évolution fatale en quelques mois.
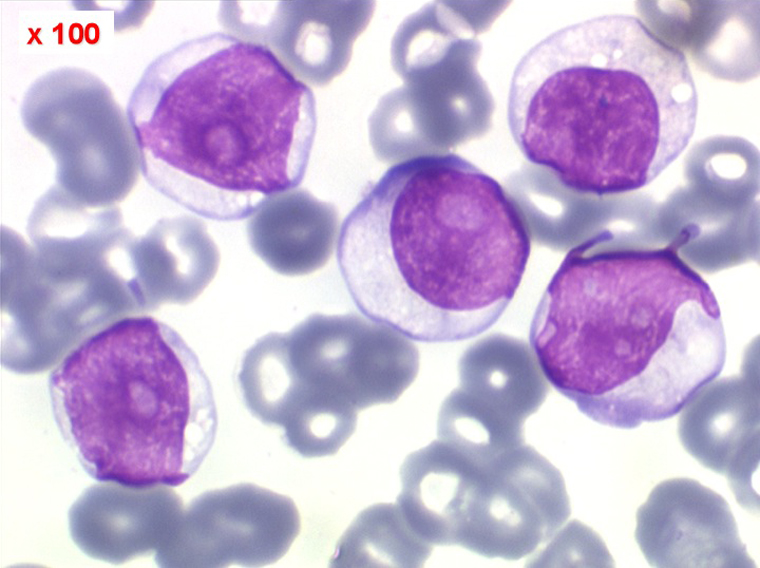
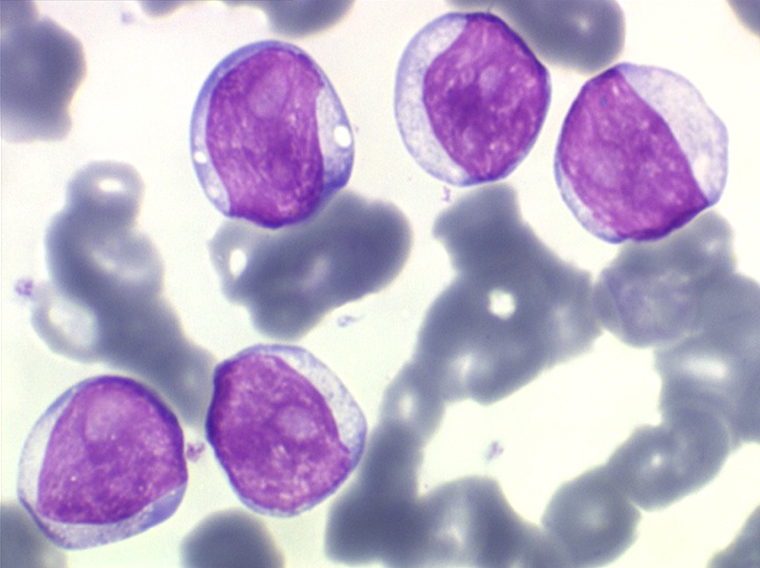
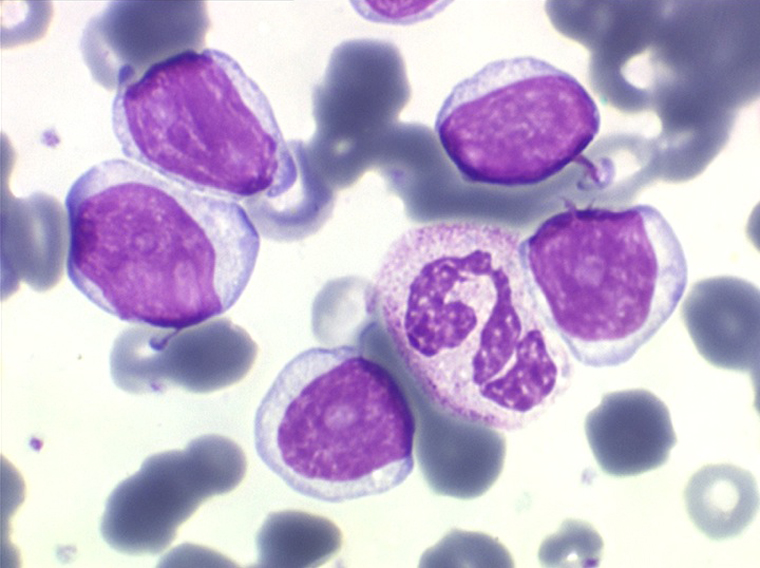
(3) – La leucémie a tricholeucocytes
La leucémie à tricholeucocytes se présente le plus souvent comme une pancytopénie avec cliniquement une splénomégalie. Une particularité de la NFS est la disparition des monocytes. Les formes « leucémiques » (hyperleucocytose) sont rares.
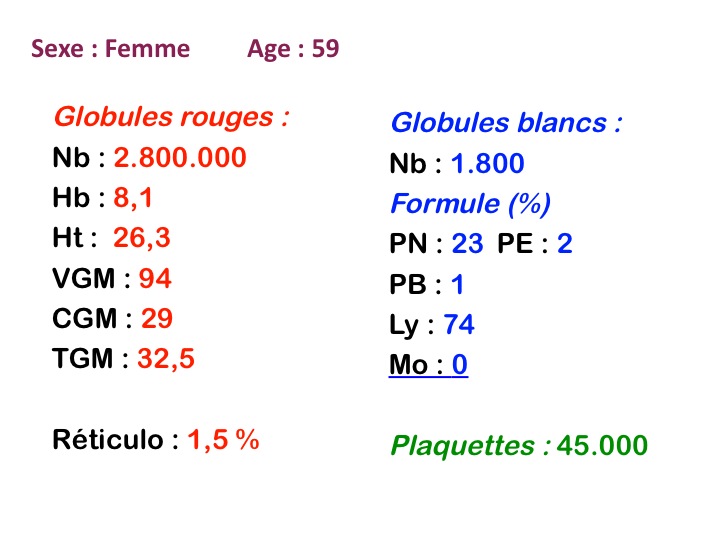
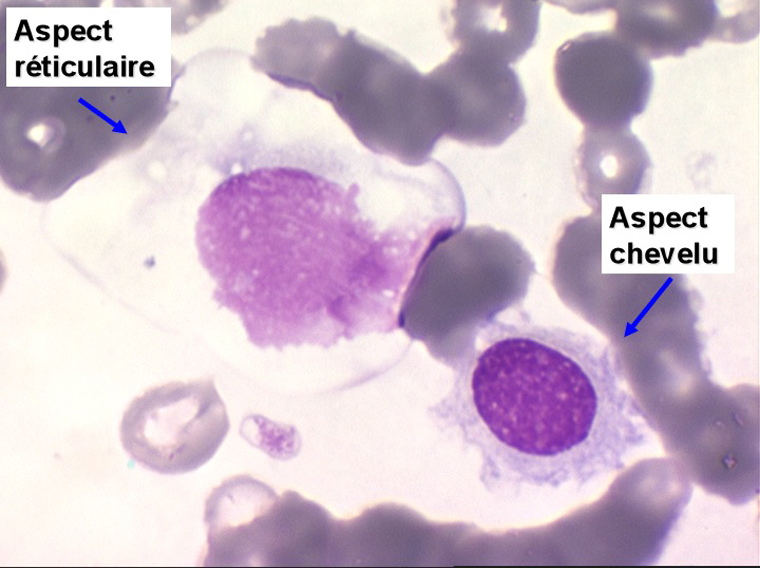
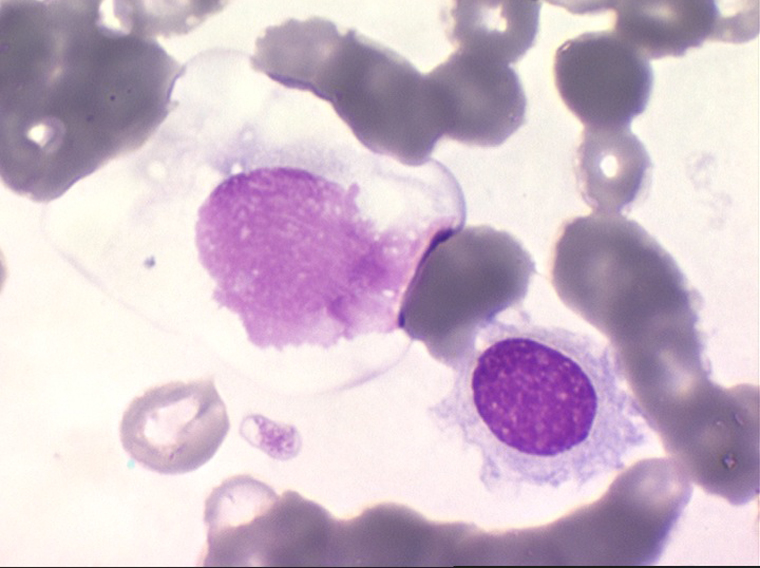
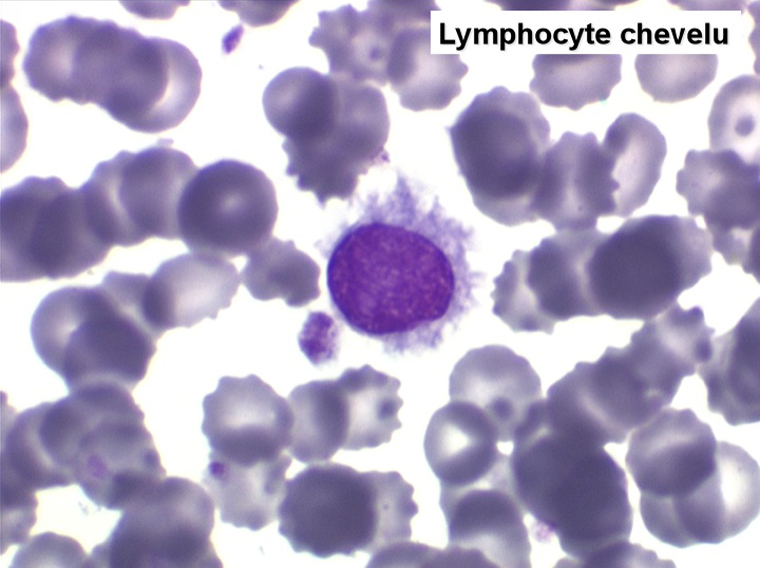
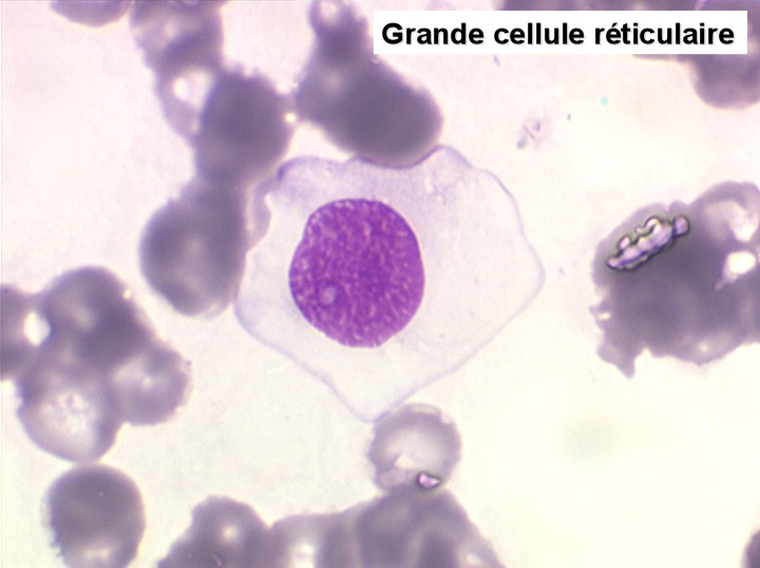
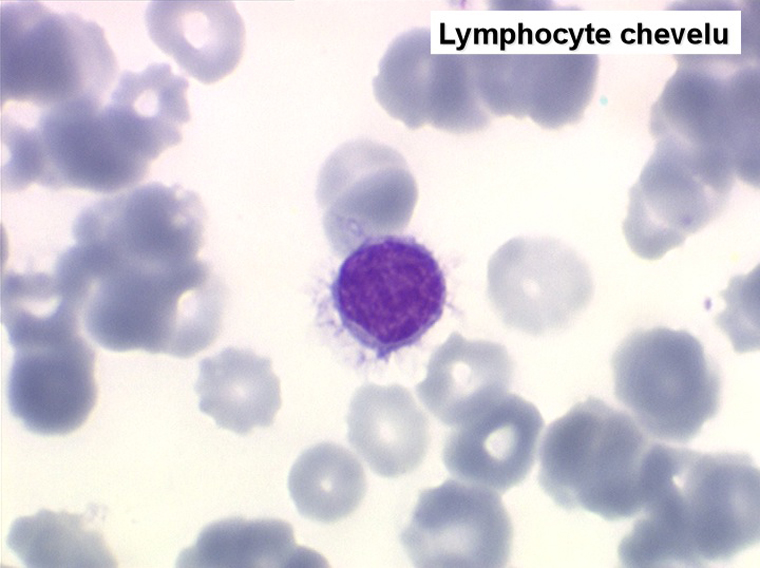
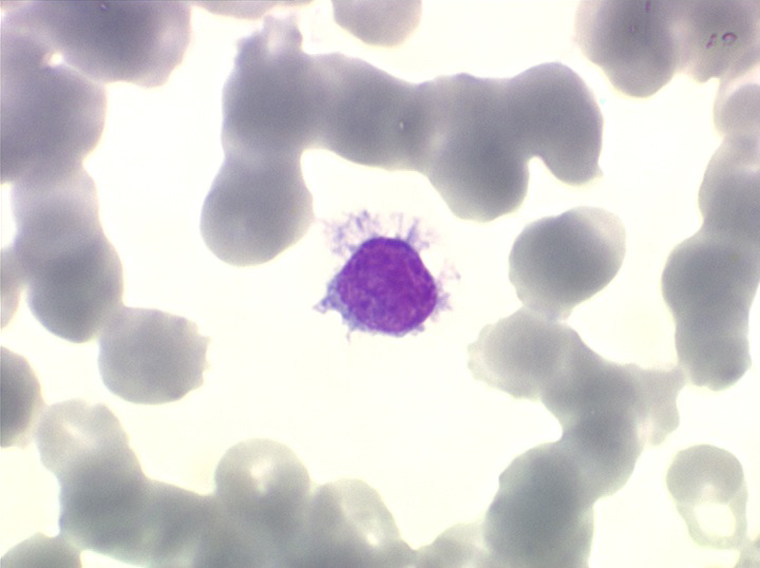
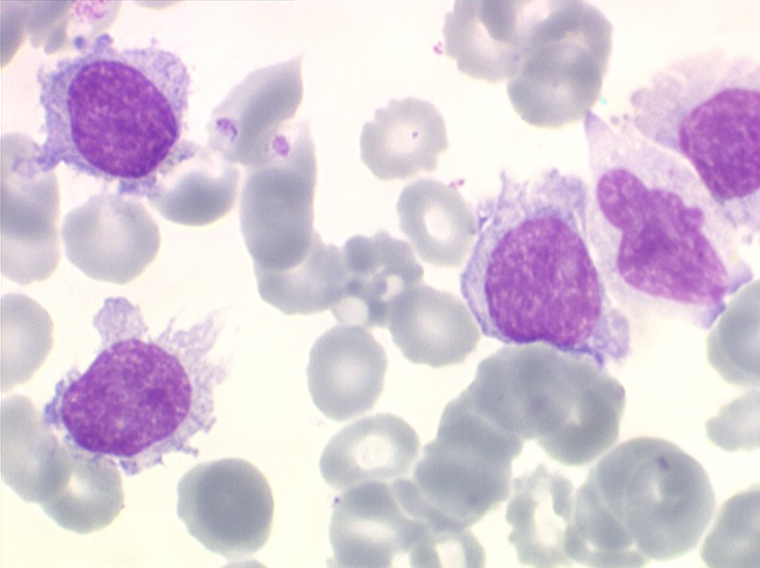
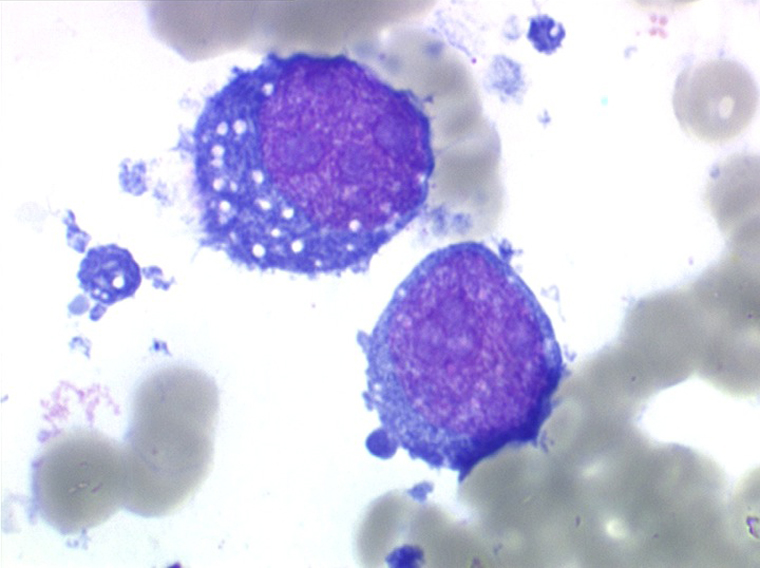
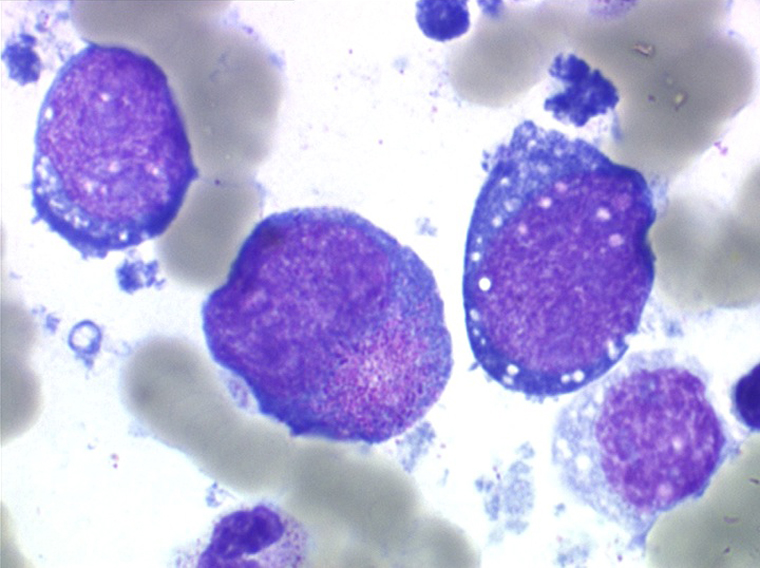
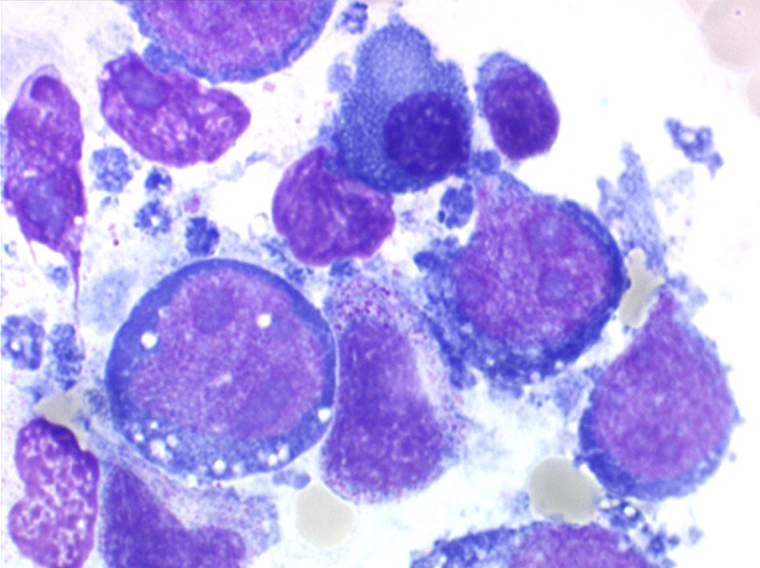
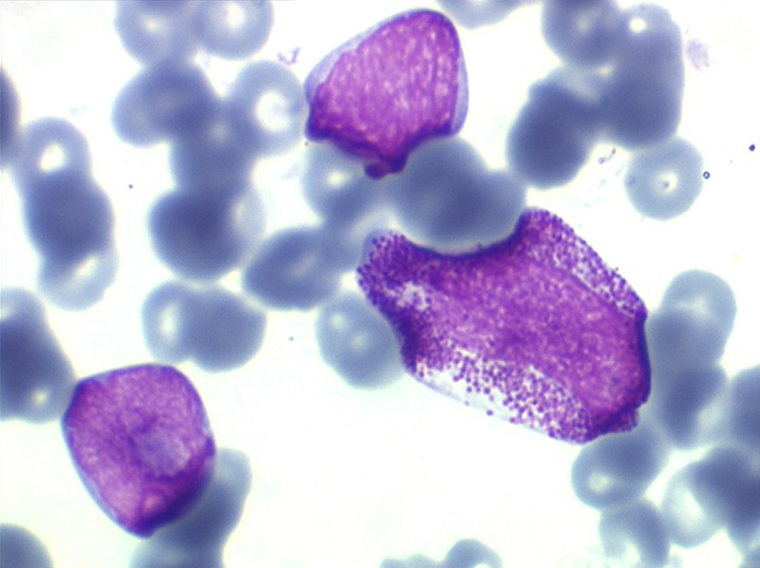
On retrouve dans le sang un pourcentage plus ou moins important (il peut y en avoir très peu) de cellules très particulières, ne ressemblant à aucune cellule sanguine normale, porteuses de marqueurs lymphoïdes, seule preuve de leur origine et qui, à l'état frais (en contraste de phase), ont une membrane filamenteuse les faisant apparaître comme chevelues (d'où leur nom). Les villosités périphériques sont très longues et occupent toute la surface membranaire.
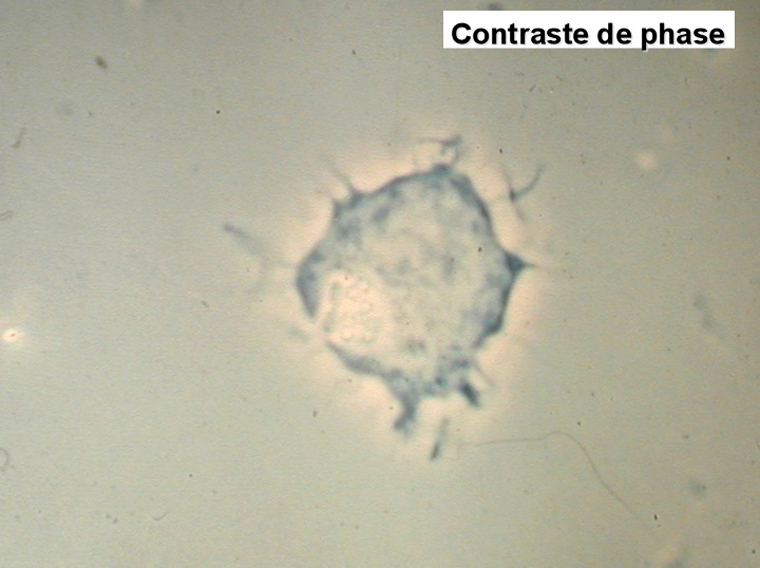
Sur les frottis colorés par le May-Grunwald-Giemsa les tricholeucocytes peuvent se présenter sous deux aspects :
• soit celui de lymphocytes chevelus, les villosités membranaires sont cependant plus courtes qu'à l'état frais.
• soit plus souvent sous forme de très grandes cellules réticulaires, à cytoplasme abondant, mal limité et optiquement vide et à noyau ovalaire, fragile, à chromatine fine, sans mottes. La cellule ressemble à un « œuf sur le plat ».
La moelle est infiltrée par ces tricholeucocytes, mais elle est surtout fibreuse, ce qui rend la ponction médullaire difficile ou improductive (intérêt de la biopsie médullaire) et explique l'insuffisance médullaire responsable de la pancytopénie avec le risque infectieux qu'elle comporte. L'évolution de cette leucémie est chronique, émaillée de complications infectieuses, mais l'intérêt de la reconnaissance de ce type de syndrome lymphoprolifératif vient de ce qu'il est sensible à des traitements spécifiques qui en améliorent considérablement le pronostic.